用于烯烃氢甲酰化合成醛和醇的多相催化剂及其制备方法与流程
本发明涉及催化剂领域,更具体地涉及一种用于烯烃氢甲酰化合成醛和醇的多相催化剂及其制备方法。
背景技术:
烯烃与合成气(CO+H2)氢甲酰化反应制取比原料烯烃高一个碳的醛的反应,是最早实现工业生产的均相催化过程,也是当今最重要的有机化工生产工艺之一。氢甲酰化产品遍及不同碳数的醛,再经简单加氢得到醇。其中最主要的是由丙烯出发氢甲酰化生产丁醛,再经缩合得到重要的增塑剂2-乙基己醇,另外以高碳烯烃(C≥6)为原料合成的高碳增塑剂醇(C8-C11)和高碳洗涤剂醇(C12-C18)占有越来越重要的地位。烯烃氢甲酰化反应最初采用可溶性的均相羰基钴(HCo(CO)4)为催化剂(F.Piacenti,M.Bianchi,P.Frediani,etal.J.Organomet.Chem.1991,(417):77-88;W.A.Herrmann,C.W.Kohlpainter.Angew.Chem.Int.Ed.Engl.,1993,(32):1524-1544)。但是HCo(CO)4稳定性差,容易分解,产品的正异比较低,须在高的一氧化碳分压下操作。采用三丁基膦配体(PBu3)改性HCo(CO)4制得的HCo(CO)3PBu3催化剂具有较高的稳定性,反应条件更为温和。目前该催化剂在高碳烯烃氢甲酰化工业生产中占有主要地位,但是存在选择性差、能耗高以及催化剂和产品分离难等问题,综合经济技术指标差。G.Wilkinson(D.Evans,J.A.Osborn,G.Wilkinson,J.Chem.Soc(A).,1968,(12):3133-3142)等发明的铑-三苯基膦催化剂(HRhCO(PPh3)3)具有更高的氢甲酰化活性、选择性和温和反应条件。然而,对于高碳烯烃氢甲酰化反应,其生成的高碳醛的沸点很高,须在高温下闪蒸才能与催化剂分离,昂贵的铑-膦催化剂在高温下遭到分解破坏,因而难以实现催化剂的循环使用,严重制约了其在烯烃氢甲酰化中的广泛应用。1984年,Ruhrchime公司和Rhone-Poulenc公司开发了两相(有机相-水相)氢甲酰化催化剂,将水溶性的HRhCO(TPPTS)3催化剂用于丙烯氢甲酰化的工业生产中,反应结束后通过简单的静止分层分离,实现了催化剂和产品的分离以及催化剂的循环使用。但是对于C≥6的高碳烯烃氢甲酰化,由于其水溶性过低而使反应受传质控制,反应活性很低,无法实现工业应用。US953016公开了在水相催化剂中添加某种促进剂,常用的促进剂包括表面活性剂、季胺盐、有机溶剂等,可以一定程度上改善溶解性,提高了活性,但是反应后产生乳化,使得催化剂和产品分离困难。CN1422695公开了一种烯烃氢甲酰化制取各类有机醛的催化剂。该催化剂由主活性组分、助剂、载体和有机配体组成。其中,主活性组分为Rh、Pt、Rh、Pd的贵金属,载体为二氧化硅、MCM-41活性炭或三氧化二铝,助剂为V、Ti、Zr、Mn等,有机配体为三苯基膦,三苯基膦三磺酸钠、三环己烷基膦、氧化三苯基膦或三酚基膦等。该催化剂为多相-均相杂化催化剂,具有多相催化剂的易分离性能与均相催化剂的高活性与高选择性。CN101642719公开了一种锚合配体修饰的负载金属催化剂及其制备方法与应用。该催化剂由载体、金属组分和有机配体组成。载体选用二氧化硅、MCM-41或SBA-15,有机配体选用含烷氧基硅烷基团的有机膦配体。在该催化剂上,有机配体和金属组分均被固定到载体上,同时有机配体与金属组分存在配位作用原位生成活性物种,该催化剂具有较高的催化活性和稳定性,且催化剂和产品分离容易。中国科学院大连化学物理研究所开发了合成气一步法制油品联产高碳直链混合伯醇的催化剂(US7670985),液体产物中油品(主要为石脑油与柴油)与高碳直链(C2~C18)混合伯醇的比例为1:1左右,具有工艺简单和经济效益高的优点。用于该工艺的催化剂中含有Co2C组分,研究发现这种Co2C具有较强的CO非解离吸附性能和CO插入性能,而有别于金属Co较强的CO解离性能,从而使得液体产物中含有一定量的醇产物。该催化剂作为合成油品联产高碳醇的催化剂,虽含有一定量的Co2C组分,但是催化剂中Co2C晶相所占比例较低,并且该专利在催化剂制备方法中未采取措施提高催化剂中Co2C晶相的含量,调控Co2C和金属Co两种晶相之间的比例。烯烃氢甲酰化反应要求催化剂具有较强的CO插入性能,从而要求催化剂含有较多的Co2C物种。因而该类催化剂不适合作为烯烃氢甲酰化的催化剂。
技术实现要素:
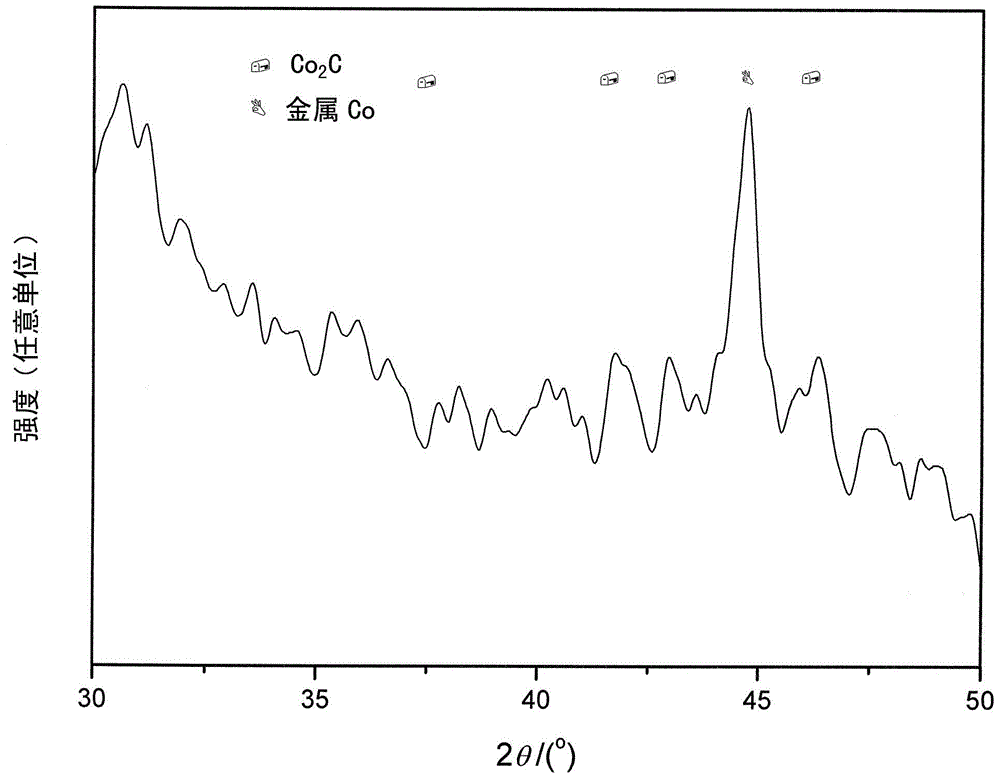
本发明利用Co2C这种具有较强的CO非解离吸附性能及CO插入性能,首次将这种含有Co2C组分的多相催化剂应用到烯烃氢甲酰化领域,并且通过在催化剂制备方法中增加碳化步骤,并优化碳化条件,使得催化剂中的金属Co部分或全部碳化为Co2C晶相,提高催化剂中Co2C的含量,调控催化剂中金属Co和Co2C两种晶相之间的比例,使得Co2C和Co复合物中Co2C的比例达到40~100%,制得高氢甲酰化性能的催化剂。另外,本发明也提供了一种活性炭负载纯相Co2C的催化剂的制备方法。本发明针对现有的烯烃均相氢甲酰化技术中存在的催化剂和产品分离难的突出问题,以及现有的多相氢甲酰化催化方法存在的氢甲酰化活性和选择性低的问题,采用活性炭负载金属Co和Co2C多相催化剂,并且通过在催化剂制备方法中增加碳化步骤,并通过调变催化剂焙烧、还原活化条件,主要是调变碳化的条件,调控催化剂中Co2C的含量,调节Co2C和金属Co两种晶相的比例,提高了催化剂催化烯烃氢甲酰化的性能。本发明的催化剂具有高的氢甲酰化活性和选择性,与产品分离容易,避免了常规采用均相催化剂所存在的催化剂和产品分离以及循环使用的问题,具有广阔的工业应用前景。本发明的目的在于提供一种烯烃氢甲酰化合成醛和醇的多相催化剂及其制备方法。本发明的又一目的在于提供上述催化剂的制备方法。为实现上述目的,在本发明的一个方面,提供了一种用于烯烃氢甲酰化合成醛和醇的多相催化剂,所述多相催化剂为负载型多相催化剂并且由主活性组分、载体和任选的助剂组成,其中所述主活性组分为Co2C或Co2C和金属Co的复合物,所述复合物中Co2C的重量含量为40~100%,所述载体为活性炭,所述助剂为选自Cu、La、Ce、Li和Sr中的一种或几种,所述烯烃为C2-C18的α烯烃。在本发明的一个优选的实施方案中,所述活性炭为杏壳活性炭或椰壳活性炭。在本发明的一个优选的实施方案中,所述活性炭的比表面积为400~2000m2/g,平均孔径为1~10nm以及孔容为0.2~2mL/g。在本发明的一个优选的实施方案中,作为所述主活性组分以Co元素计的重量含量为所述多相催化剂重量的5~20%。在本发明的一个优选的实施方案中,所述助剂的重量含量为所述多相催化剂重量的0.01~3%。在本发明的另一个方面,提供了一种制备上述多相催化剂的方法,所述方法包括以下步骤:(1)采用浸渍法将所述主活性组分的可溶性无机盐水溶液和任选的助剂的可溶性无机盐水溶液浸渍在活性炭载体上并烘干,得到催化剂无机盐前驱物;(2)将所述催化剂无机盐前驱物装填在固定床反应器,原位依次进行干燥、焙烧、还原活化和碳化反应,制得催化剂,其中在惰性气氛下干燥和焙烧,得到催化剂氧化物前驱物;将所述催化剂氧化物前驱物还原活化成含金属Co的催化剂金属态前驱体;将所述催化剂金属态前驱体进行碳化以使金属Co全部或部分碳化为Co2C,并且使Co2C和Co复合物中Co2C的重量含量为40~100%(例如通过调变催化剂焙烧、还原活化条件,主要是调变碳化的条件,调控催化剂中Co2C的含量,调节Co2C和金属Co两种晶相的比例),从而得到所述多相催化剂。在本发明的一个优选的实施方案中,在进行浸渍之前,所述活性炭载体经过去离子水煮沸洗涤处理。在本发明的一个优选的实施方案中,在进行焙烧之前,所述催化剂无机盐前驱物在惰性气氛下进行干燥以除去残留水分,其中干燥所用的压力为0.1~1.0MPa,温度为373~393K,空速为300~2000h-1,时间为2~20小时。在本发明的一个优选的实施方案中,步骤(1)中的烘干在323~343K温度下的空气气氛中进行12~36小时。在本发明的一个优选的实施方案中,步骤(2)中的焙烧所用的压力为0.1~1.0MPa,温度为473~773K,空速为300~2000h-1,时间为4~24小时。在本发明的一个优选的实施方案中,步骤(2)中的还原所用的还原气体为氢气含量为10~100%的含氢混合气,其中所述含氢混合气中除氢气以外的其他气体为氮气或氩气;并且还原所用的温度为573~773K,压力为0.1~1.0MPa,空速为300~2000h-1,时间为2~48小时。在本发明的一个优选的实施方案中,步骤(4)中的碳化所用的气体为CO含量为5~100%的含CO气体,所述含CO气体中除CO以外的气体为氢气、氮气或氩气;并且碳化所用的温度为463~513K,压力为0.1~10.0MPa,空速为100~2000h-1,时间为2~500小时。在本发明的一个优选的实施方案中,所述干燥、焙烧、还原和碳化在固定床反应器中原位进行。本发明的催化剂是一种负载型多相催化剂,具有较高的氢甲酰化活性和选择性,而且其和产品分离容易,避免了常规采用均相催化剂所存在的催化剂和产品分离以及循环使用的问题,具有广阔的工业应用前景。附图说明图1为根据本发明实施例制备的催化剂的XRD谱图。图2为根据本发明实施例制备的催化剂L的XRD谱图具体实施方式本发明提供了一种用于烯烃氢甲酰化合成醛和醇的多相催化剂及其制备方法。烯烃氢甲酰化反应需要催化剂活性位上具有较强的CO非解离吸附活化和CO插入到烯烃分子中进行反应的性能。烯烃氢甲酰化反应和合成气(CO+H2)一步法制油品联产高碳直链混合伯醇反应在CO非解离吸附活化以及CO插入到烯烃分子中进行反应方面基本一致。本发明的发明人利用Co2C上CO较强的非解离吸附活化和CO插入性能,首次将这种含有Co2C组分的多相催化剂应用到烯烃氢甲酰化领域,并且通过在催化剂制备方法中增加碳化步骤,使得催化剂中的金属Co部分或全部碳化为Co2C晶相。通过调变、优化催化剂焙烧、还原活化条件,主要是调变、优化碳化的条件,调控催化剂中Co2C的含量,调节Co2C和金属Co两种晶相之间的比例,使得Co2C和金属Co复合物中Co2C的比例控制在40~100%,制得具有优异性能的氢甲酰化催化剂。为此,本发明提供一种烯烃氢甲酰化合成醛和醇的多相催化剂,该催化剂为负载型多相催化剂,由主活性组分和载体组成,或由主活性组分、助剂及载体组成。催化剂中主活性组分为Co2C或为Co2C和金属Co的复合物,载体为活性炭。当催化剂中含有助剂时,助剂为Cu、La、Ce、Li和Sr中的一种或几种,其中所述烯烃为碳数2~18的α烯烃。在本发明的一个优选的实施方案中,催化剂由主活性组分和载体组成。在本发明的另一个优选的实施方案中,催化剂由主活性组分、助剂及载体组成。优选地,载体为杏壳活性炭或椰壳活性炭。优选地,活性炭的比表面积为400~2000m2/g,平均孔径为1~10nm,孔容为0.2~2mL/g。在本发明的另一个优选的实施方案中,主活性组分为Co2C,在本发明的另一个更优选的实施方案中,为Co2C和金属Co的复合物,Co2C和金属Co两种晶相的比例可控。优选地,所述活性组分Co2C和金属Co重量含量(以Co元素为计算基准)为催化剂重量的5~20%。优选地,当催化剂中添加助剂时,助剂含量为催化剂重量0.01~3%。在本发明的另一方面,提供一种制备如上所述的催化剂的方法,所述方法包括以下步骤:(1)采用浸渍法将主活性组分或主活性组分及助剂的可溶性无机盐的水溶液浸渍在活性炭载体上并烘干,以制得催化剂无机盐前驱物。优选地,其中的烘干在323~343K温度下在空气气氛中进行12~36小时。任选地,在该步骤(1)之前,使活性炭经过去离子水煮沸洗涤处理。(2)将所述催化剂无机盐前驱物装填在固定床反应器,原位依次进行干燥、焙烧、还原活化和碳化反应,制得催化剂。需要说明的是,为了获得性能更优异的多相催化剂,在制备过程中,优选地对活性炭载体进行选择和上述水洗涤处理。并且,通过调变催化剂焙烧、还原活化条件,主要是调变碳化的条件,调控催化剂中Co2C的含量,调节Co2C和金属Co两种晶相的比例,从而得到所述多相催化剂。由于Co2C和金属Co晶相的准确含量以及各自所占比例很难测定,以Co2C和金属Co衍射峰的峰高比例来表示两种晶相的所占比例。优选地,对活性炭载体进行选择和上述水洗涤处理。而且,催化剂干燥、焙烧、还原活化和碳化过程中的条件如温度、压力、空速、时间、气氛等优选处于上述范围内。另外,在催化剂中添加助剂时,可促进碳化生成Co2C活性位。优选地,具体步骤如下:在惰性气氛下干燥和焙烧,得到催化剂氧化物前驱物。优选地,焙烧所用的气体为氮气或氩气惰性气体,压力为0.1~1.0MPa,温度为473~773K,空速为300~2000h-1,时间为4~24小时。任选地,在该步骤(2)之前,所述催化剂无机盐前驱物在惰性气氛(例如氮气或氩气)下干燥以除去残留水分,并且优选地,干燥所用的气体为氮气或氩气惰性气体,压力为0.1~1.0MPa,温度为373~393K,空速为300~2000h-1,时间为2~20小时。将上述催化剂氧化物前驱物还原活化成含金属Co的催化剂金属态前驱体。优选地,还原所用的还原气体为含氢混合气,氢含量为10~100%,含氢混合气中氢气以外的其他气体为氮气或氩气,并且还原所用的温度为573~773K,压力为0.1~1.0MPa,空速为300~2000h-1,时间为2~48小时。将上述金属态催化剂前驱体进行碳化,使得金属Co全部或部分碳化为Co2C,从而使得催化剂中主活性组分为Co2C或为Co2C和金属Co的复合物,由此制备得到所述多相催化剂。优选地,碳化所用的气体为含CO气体,CO含量为5~100%,含CO气体中其它以外的为氢气、氮气或氩气,并且碳化所用的温度为463~513K,压力为0.1~10.0MPa,空速为100~2000h-1,时间为2~500小时。优选地,上述催化剂的干燥、焙烧、还原和碳化过程在氢甲酰化固定床反应器中原位进行。在本发明的催化剂中,经过一系列步骤制备得到的多相Co2C拥有的较强的CO非解离吸附以及CO线性插入的性能。而CO非解离吸附以及CO线性插入正是烯烃氢甲酰化反应的关键的步骤,因而,本发明的催化剂可催化烯烃氢甲酰化反应。在该催化剂中Co2C活性位或Co2C和金属Co复合物活性位催化作用下,烯烃氢甲酰化生成醛,其中部分醛加氢生成醇,形成醛和醇的混合物。该混合物只需再经简单加氢合成目标产物醇。实施例下面通过具体实施例对本发明做进一步说明。除非另有具体说明,本申请中所用的“百分比”和“份”都基于重量。在下面的实施例中,所有的原料如下:椰壳活性炭:唐山联合炭业有限公司;杏壳活性炭:唐山联合炭业有限公司;Co(NO3)2·6H2O:中国医药(集团)上海化学试剂公司,分析纯;Cu(NO3)2·3H2O:中国医药(集团)上海化学试剂公司,分析纯;La(NO3)3·6H2O:中国医药(集团)上海化学试剂公司,分析纯;Ce(NO3)3·6H2O:中国医药(集团)上海化学试剂公司,分析纯;Li(NO3)3:中国医药(集团)上海化学试剂公司,分析纯;Sr(NO3)2:中国医药(集团)上海化学试剂公司,分析纯;Zr(NO3)4·5H2O:中国医药(集团)上海化学试剂公司,分析纯;SiO2:青岛海洋化工厂,纯度≥99.5%;H2:大连大特气体有限公司,纯度≥99.999体积%;N2:大连大特气体有限公司,纯度≥99.999体积%;CO:大连大特气体有限公司,纯度≥99.999体积%;CO/Ar混合气(含有30体积%H2、70体积%CO):大连大特气体有限公司,纯度≥99.999体积%;H2/CO混合气(含有50体积%H2、50体积%CO):大连大特气体有限公司,纯度≥99.999体积%;H2/CO混合气(含有20体积%H2、80体积%CO):大连大特气体有限公司,纯度≥99.999体积%;H2/CO混合气(含有66.6体积%H2、33.4体积%CO):大连大特气体有限公司,纯度≥99.999体积%。实施例1椰壳活性炭的比表面积和孔径分布测定在QUANTACHROME公司的AS-1型吸附仪上进行。样品先在623K下脱气处理3h,在液氮温度下进行氮吸附测试。用BET法计算样品的比表面积,用BJH法计算孔分布。所测得的杏壳活性炭比表面积为1056m2/g,平均孔径为3.4nm,孔容为0.45mL/g。取1000克椰壳活性炭载体,采用去离子水煮沸洗涤方法进行处理6次,具体方法为:取1000克椰壳活性炭,加入8000克去离子水,共煮沸0.5小时,随后将活性炭与沸水分离,再次加入去离子水进行下一次煮沸洗涤。洗好的活性炭在393K温度下干燥12小时,标记为AC1,备用。采用以上方法测定标记为AC2的活性炭的比表面积为1082m2/g,平均孔径为3.5nm,孔容为0.46mL/g。按照下述步骤制备催化剂A:以20~40目处理过的椰壳活性炭为载体。称取10克处理过的活性炭。称取8.7克Co(NO3)2·6H2O,溶解于10mL去离子水中制成溶液。用常规浸渍法将制得的溶液担载在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在333K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气压力为0.1MPa,空速为500h-1;随后以1K/min速率升温至623K,在氮气气氛中该温度下焙烧8小时,氮气压力为0.1MPa,空速为500h-1(得到催化剂氧化物前驱物);温度降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为1000h-1,以1K/min速率升温至703K,在703K下还原10小时(得到催化剂金属态前驱体);温度降至373K,切换CO气体进行碳化,CO压力为0.1MPa,CO空速为100h-1,以1K/min速率升温至493K,在493K下碳化400小时,制得催化剂A(即得到本发明的多相催化剂)。采用XRD检测催化剂中Co存在的晶相,XRD测试在PANalytical公司X’PertPRO型X射线衍射仪上进行,测试条件为:Cukα1辐射源,管压40KV,管流40mA,扫描范围为2θ=5°-75°,扫描速度为0.2°/s。测得的结果如图1中的谱图(6)所示。由谱图(6)可见,2θ=37.0、41.3、42.6和45.8系列衍射峰为Co2C晶相的衍射峰,其中42.6处为主衍射峰,表明催化剂A中仅含有Co2C晶相,即催化剂A中主活性组分Co2C的含量为100%。其重量含量为制备催化剂加入的金属Co量,为15%,该多相催化剂标记为15Co2C(100)/AC1。实施例2以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂B:称取8.7克Co(NO3)2·6H2O,溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在353K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中393K温度下烘干4小时,氮气压力为0.1MPa,空速为500h-1;随后升温至573K温度下在氮气气氛中焙烧12小时,升温速率为1K/min,氮气压力为0.1MPa,氮气空速为300h-1;温度降至373K,切换H2进行还原活化,H2压力为0.5MPa,空速为500h-1,以1K/min速率升温至683K,在683K下还原20小时;温度降至373K,切换CO气体进行碳化,CO压力为0.1MPa,CO空速为100h-1,以1K/min速率升温至493K,在493K下碳化50小时,制得催化剂B。与实施例1类似,采用XRD检测催化剂B中Co存在的晶相,结果如图1中的谱图(8)所示。从谱图(8)可见,在2θ=44.2°出现归属于金属Co晶相结构的衍射峰,在2θ=37.0、41.3、42.6和45.8出现归属于Co2C晶相结构的衍射峰,表明催化剂B中含有Co2C和金属Co的晶相,即催化剂B的主活性组分为Co2C和金属Co的复合物。其中,2θ=42.6处Co2C主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为40%和60%。以Co2C和金属Co衍射峰高的比例表示两种晶相所占比例的相对含量及其变化趋势,并通过所占比例的变化与催化剂性能相关联,下同。两种晶相按Co计的重量含量为15%。将该多相催化剂标记为15Co2C(40)@Co(60)/AC1。实施例3以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂C:称取8.7克Co(NO3)2·6H2O,溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在353K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气压力为0.3MPa,氮气空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧12小时,升温速率为1K/min,氮气压力为0.3MPa,空速为300h-1;温度降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为500h-1,升温速率为1K/min,在703K下还原10小时;温度降至373K,切换H2/CO混合气(含有50体积%H2、50体积%CO)进行碳化,H2/CO混合气压力为3.0MPa,空速为500h-1,以1K/min速率升温至493K,在493K下碳化50小时,制得催化剂C。与实施例1类似,采用XRD检测催化剂C中Co存在的晶相,结果如图1中的谱图(7)所示。从谱图(7)可见,催化剂C中含有Co2C和金属Co的晶相,即催化剂C的主活性组分为Co2C和金属Co的复合物,其中,2θ=42.6处Co2C的主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为50%和50%。它们按Co计的重量含量为15%。将该多相催化剂标记为15Co2C(50)@Co(50)/AC1。实施例4以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂D:称取8.7克Co(NO3)2·6H2O,将它们溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用等体积浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在353K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中393K温度下烘干4小时,氮气空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧12小时,升温速率为1K/min,氮气空速为300h-1;降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为500h-1,升温速率为1K/min,在653K下还原20小时;降至373K,切换CO/Ar混合气(含有30体积%CO、70体积Ar%)进行碳化,CO/Ar混合气压力为0.5MPa,空速为300h-1,升温速率为2K/min,在473K下碳化100小时,制得催化剂D。与实施例1类似,采用XRD检测催化剂D中Co存在的晶相,结果如图1中的谱图(1)所示。从谱图(1)可见,催化剂E中含有Co2C和金属Co的晶相,即催化剂D的主活性组分为Co2C和金属Co的复合物,其中,谱图中2θ=42.6处Co2C主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为55%和45%。它们按Co计的重量含量为15%。将该多相催化剂标记为15Co2C(55)@Co(45)/AC1。实施例5以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂E:称取5.48克Co(NO3)2·6H2O,溶解于12mL去离子水中制成溶液。称取10克处理过的活性炭,采用常规浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在343K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧12小时,升温速率为1K/min,氮气空速为300h-1(得到催化剂氧化物前驱物);降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为500h-1,升温速率为1K/min,在673K下还原20小时(得到催化剂金属态前驱物);降至373K,切换H2/CO混合气(含有50体积%H2、50体积%CO)进行碳化,H2/CO混合气压力为3.0MPa,空速为500h-1,升温速率为1K/min,在493K下碳化2小时,制得制得催化剂E(即得到本发明的多相催化剂)。与实施例1类似,采用XRD检测催化剂C中Co存在的晶相(谱图未显示),结果为催化剂E中含有Co2C和金属Co的晶相,即催化剂E的主活性组分为Co2C和金属Co的复合物,Co2C主衍射峰高与金属Co衍射峰高的比例分别为45%和55%,它们按Co计的重量含量为10%,将该多相催化剂标记为10Co2C(45)@Co(55)/AC1。实施例6以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂F:称取12.49克Co(NO3)2·6H2O和0.48克Cu(NO3)2·3H2O,将它们溶解于8mL去离子水中制成溶液。称取10克处理过的活性炭,采用常规浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在343K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧12小时,升温速率为2K/min,氮气空速为300h-1;降至373K,切换H2进行还原活化,H2压力为0.1MPa,H2空速为500h-1,升温速率为1K/min,在673K下还原20小时;降至373K,切换H2/CO混合气(含有66.6体积%H2、33.4体积%CO)进行碳化,H2/CO混合气压力为4.0MPa,空速为300h-1,升温速率为2K/min,在493K下碳化10小时,制得催化剂F。与实施例1类似,采用XRD检测催化剂F中Co存在的晶相,结果如图1中的谱图(2)所示。从谱图(2)可见,催化剂F中含有Co2C和金属Co的晶相,即催化剂F的主活性组分为Co2C和金属Co的复合物,其中,2θ=42.6处Co2C衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为75%和25%。它们按Co计的重量含量为20%。将该多相催化剂标记为20Co2C(75)@Co(25)1Cu/AC1。实施例7以实施例1中处理好的椰壳活性炭为载体,按照下述步骤制备催化剂G:称取4.61克Co(NO3)2·6H2O和0.111克La(NO3)3·6H2O,将它们溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液浸渍在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在333K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧12小时,升温速率为1K/min,氮气空速为300h-1;降至373K,切换H2进行还原活化,H2压力为0.5MPa,H2空速为500h-1,升温速率为2K/min,在703K下还原10小时;降至373K,切换H2/CO混合气(含有60体积%CO、40体积H2%)进行碳化,H2/CO混合气压力为3.0MPa,空速为500h-1,升温速率为2K/min,在493K下碳化40小时,制得催化剂G。与实施例1类似,采用XRD检测催化剂G中Co存在的晶相,结果如图1中的谱图(3)所示。从谱图(3)可见,催化剂G中含有Co2C和金属Co的晶相,即催化剂G的主活性组分为Co2C和金属Co的复合物,其中,2θ=42.6处Co2C主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为60%和40%。它们按Co计的重量含量为8%。将该多相催化剂标记为8Co2C(60)@Co(40)0.3La/AC1。实施例8杏壳活性炭的比表面积和孔径分布测定在QUANTACHROME公司的AS-1型吸附仪上进行。样品先在623K下脱气处理3h,在液氮温度下进行氮吸附测试。用BET法计算样品的比表面积,用BJH法计算孔分布。所测得的杏壳活性炭比表面积为765m2/g,平均孔径为3.6nm,孔容为0.65mL/g。取1000克杏壳活性炭载体,采用去离子水煮沸洗涤方法进行处理6次,具体方法为:取1000克杏壳活性炭,加入8000克去离子水,共煮沸0.5小时,随后将活性炭与沸水分离,再次加入去离子水进行下一次煮沸洗涤。洗好的活性炭在393K温度下干燥12小时,标记为AC2,备用。采用以上方法测定AC2活性炭比表面积为781m2/g,平均孔径为3.7nm,孔容为0.66mL/g。以20~40目处理过的杏壳活性炭为载体,按照下述步骤制备催化剂H:称取8.7克Co(NO3)2·6H2O,溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液担载在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在333K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中383K温度下烘干6小时,氮气压力为0.1MPa,空速为500h-1;随后升温至623K,在氮气气氛中该温度下焙烧8小时,升温速率为1K/min,氮气压力为0.1MPa,空速为500h-1(得到催化剂氧化物前驱物);温度降至373K,切换H2进行还原活化,H2压力为1.0MPa,空速为500h-1,升温速率为2K/min,在703K下还原10小时(得到催化剂金属态前驱体);温度降至373K,切换H2/CO混合气(含有50体积%CO、50体积%H2)进行碳化,H2/CO混合气的压力为3.0MPa,空速为300h-1,升温速率为2K/min,在493K下碳化50小时,制得催化剂H。与实施例1类似,采用XRD检测催化剂H中Co存在的晶相,结果如图1中的谱图(4)所示。从谱图(4)可见,催化剂I中含有Co2C和金属Co的晶相,即催化剂H的主活性组分为Co2C和金属Co的复合物。其中,2θ=42.6处Co2C主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为50%和50%。它们按Co计的重量含量为15%。将该多相催化剂标记为15Co2C(50)@Co(50)/AC2。实施例9以20~40目处理过的杏壳活性炭为载体,按照下述步骤制备催化剂I:称取2.59克Co(NO3)2·6H2O,溶解于14mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液担载在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在343K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在383K温度下烘干6小时,氮气压力为0.1MPa,空速为500h-1;随后升温至623K,在氮气气氛中该温度下焙烧8小时,升温速率为2K/min,氮气压力为0.1MPa,空速为500h-1;温度降至373K,切换H2进行还原活化,H2压力为1.0MPa,空速为500h-1,升温速率为2K/min,在703K下还原10小时;温度降至373K,切换H2/CO混合气(含有50体积%CO、50体积H2%)进行碳化,H2/CO混合气的压力为3.0MPa,空速为300h-1,升温速率为2K/min,在493K下碳化50小时,制得催化剂I。与实施例1类似,采用XRD检测催化剂I中Co存在的晶相,结果如图1中的谱图(9)所示。从谱图(9)可见,催化剂I中含有Co2C和金属Co的晶相,即催化剂I的主活性组分为Co2C和金属Co的复合物。从谱图(9)中发现,Co2C和金属Co衍射峰比较弱,也表明钴负载量低,Co2C和金属Co呈高分散状态。Co2C主衍射峰高与金属Co衍射峰高的比例分别为55%和45%。它们按Co计的重量含量为5%,将该多相催化剂标记为5Co2C(55)@Co(45)/AC2。实施例10以20~40目处理过的杏壳活性炭为载体,按照下述步骤制备催化剂J:称取12.65克Co(NO3)2·6H2O和0.155克Sr(NO3)3溶解于8mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液担载在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在343K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧、还原活化以及碳化步骤。具体条件为:在氮气气氛中在383K温度下烘干6小时,氮气压力为0.1MPa,空速为500h-1;随后升温至573K,在氮气气氛中该温度下焙烧10小时,升温速率为2K/min,氮气压力为0.1MPa,空速为500h-1;温度降至373K,切换H2进行还原活化,H2压力为1.0MPa,空速为500h-1,升温速率为1K/min,在703K下还原15小时;温度降至373K,切换H2/CO混合气(含有50体积%CO、50体积%H2)进行碳化,H2/CO混合气的压力为3.0MPa,空速为500h-1,升温速率为2K/min,在493K下碳化30小时,制得催化剂J。与实施例1类似,采用XRD检测催化剂J中Co存在的晶相(谱图未显示),结果为催化剂J中含有Co2C和金属Co的晶相,即催化剂J的主活性组分为Co2C和金属Co的复合物,Co2C主衍射峰高与金属Co衍射峰高的比例分别为80%和20%,它们按Co计的重量含量为20%,并且作为助剂的Sr含量为0.5%,将该多相催化剂标记为20Co2C(80)@Co(20)0.5Sr/AC2。对比例1以以下处理好的杏壳活性炭为载体,催化剂按照下述步骤制备催化剂K。取1000克杏壳活性炭载体,先采用0.5%(wt)稀盐酸煮沸洗涤,再用去离子水煮沸洗涤方法处理6次。具体方法为:取1000克杏壳活性炭,加入8000克0.5%(wt)稀盐酸,煮沸0.5小时,随后将活性炭与热稀盐酸水溶液分离,加入去离子水进行煮沸洗涤6次。洗好的活性炭在393K温度下干燥12小时,标记为AC3,备用。采用以上实施例1中的方法测定AC3活性炭,其比表面积为796m2/g,平均孔径为3.9nm,孔容为0.68mL/g。以20~40目AC3为载体,按照下述步骤制备催化剂K:称取8.7克Co(NO3)2·6H2O,溶解于10mL去离子水中制成溶液。称取10克处理过的活性炭,用常规浸渍法将制得的溶液担载在活性炭上,即在室温下,将活性炭浸泡在制得的溶液中,搅拌1~2分钟,使得溶液均匀浸渍在活性炭载体上。在空气气氛中在333K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干、焙烧以及还原活化步骤。具体条件为:在氮气气氛中393K温度下烘干4小时,氮气压力为0.1MPa,空速为500h-1;随后升温至623K,在氮气气氛中该温度下焙烧8小时,升温速率为1K/min,氮气压力为0.1MPa,空速为500h-1;温度降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为1000h-1,升温速率为1K/min,在703K下还原10小时,得到催化剂K。与实施例1类似,采用XRD检测催化剂K中Co存在的晶相,结果如图1中的谱图(5)所示。从谱图(5)可见,催化剂K中仅含有金属Co的晶相,即催化剂K的主活性组分为金属Co,其重量含量为15%,将该催化剂标记为15Co(100)/AC3。对比例2以实施例8中处理好的杏壳活性炭为载体,按照专利US7670985制备催化剂L,步骤如下。称取8.75克Co(NO3)2·6H2O和0.28克Zr(NO3)4·5H2O溶解于10mL去离子水中制成溶液,并置于分液漏斗中,中间开孔的橡皮塞将分液漏斗与锥形抽滤瓶密封连接。用真空浸渍法将制得的溶液浸渍在活性炭上,即在室温下,称取10克处理过的活性炭放入锥形抽滤瓶中,将活性炭在真空(0.01MPa)下处理半小时后,打开活塞将上述制得的溶液迅速流入抽滤瓶中将活性炭载体浸泡3~5分钟。室温下阴干,在空气气氛中在333K温度下干燥24小时,制得催化剂无机盐前驱物。取4mL催化剂无机盐前驱物装填在氢甲酰化反应的固定床反应器中,原位依次进行烘干和还原活化。具体条件为:在氮气气氛中在393K温度下烘干4小时,氮气压力为0.1MPa,氮气空速为500h-1;温度降至373K,切换H2进行还原活化,H2压力为0.1MPa,空速为500h-1,升温速率为1K/min,在673K下还原6小时,制得催化剂L。与实施例1类似,采用XRD检测催化剂L中Co存在的晶相,结果如图2中的谱图所示。从谱图可见,催化剂L中含有Co2C和金属Co的晶相,即催化剂L的主活性组分为Co2C和金属Co的复合物,其中,2θ=42.6处Co2C的主衍射峰高与2θ=44.2处金属Co衍射峰高的比例分别为15%和85%。它们按Co计的重量含量为15%。将该多相催化剂标记为15Co2C(15)@Co(85)0.5La/AC2。应用评价将催化剂A-L在氢甲酰化固定床反应器中原位处理后,温度降至453K,切换H2/CO混合气(含有50体积%H2、50体积%CO),在温度为453K,反应压力为3.0MPa,气体空速为500h-1、1-己烯液体空速为0.6h-1的条件下连续进料进行反应,催化剂A-L的组成和和评价结果列于表1。由表1可见,只含有Co2C作为主活性组分的催化剂A的1-己烯转化率为6.6%,加氢生成正己烷的选择性为33.6%,异构化生成2,3-己烯的选择性为49.3%,氢甲酰化生成庚醛(包括正庚醛与异庚醛)的选择性4.3%,其中醛的正异比为3左右,生成庚醇(包括正庚醇与异庚醇)的选择性为12.8%,其中醇的正异比为5左右。以Co2C和Co的复合物作为主活性组分、椰壳炭为载体的催化剂B-G的氢甲酰化活性较高,生成庚醛和庚醇的选择性也高于催化剂A的选择性;以Co2C和Co的复合物作为主活性组分、杏壳炭为载体的催化剂H-J的氢甲酰化活性较低,生成庚醛和庚醇的选择性稍低于催化剂A-G生成庚醛和庚醇的选择性。表1:催化剂(A-L)多相催化1-己烯氢甲酰化合成庚醛和庚醇反应性能反应条件:温度:453K;压力3.0MPa;H2/CO混合气(含有50体积%H2、50体积%CO)空速为500h-1、1-己烯液体进料空速为0.6h-1;反应时间:12小时。对比例1中的催化剂K(只含有金属Co作为主活性组分)的氢甲酰化结果显示,其氢甲酰化生成醛和醇的选择性很低,分别为2.9%和1.1%。采用专利7670985制备的对比例2中催化剂L含有少量的Co2C(Co2C与金属Co复合物中Co2C的比例只有15%),氢甲酰化结果显示,生成醛和醇的选择性分别为4.0%和2.1%。综合催化剂XRD谱图和表1的反应数据,可以看出,随着催化剂中Co2C所占比例的增加,催化剂催化1-己烯氢甲酰化反应的活性和选择性表现出增加的趋势。随着催化剂中Co2C所占比例进一步的增加,催化剂的活性和选择性有所下降。只含有纯相Co2C的催化剂具有一定的氢甲酰化性能,只含有纯相金属Co的催化剂的氢甲酰化花性能很低,催化剂同时含有Co2C和金属Co,且Co2C所占比例较高,Co2C和金属Co两种晶相达到合适的比例,具有较高的氢甲酰化性能。本发明通过调变、优化催化剂焙烧、还原活化条件,增加催化剂碳化步骤,并通过调变、优化碳化的条件,调控催化剂中Co2C的含量,调节Co2C和金属Co两种晶相之间的所占比例,制得具有优异性能的氢甲酰化催化剂。本发明的催化剂是一种多相催化剂,克服了传统的多相催化剂氢甲酰化性能低的问题,可用于C2~C18的α烯烃的氢甲酰化反应,具有高活性、高选择性和高稳定性能的优点,而且实现了反应产品与催化剂的简单分离,解决了烯烃氢甲酰化采用常规匀相催化过程所存在的液体产品与均相催化剂分离难的问题,具有广阔的工业应用前景。应当指出,对于使本技术领域的专业技术人员,在不脱离本发明技术原理的前提下,是能够实现对这些实施例的多种修改的,而这些修改也应视为本发明应该保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1