负载金属催化剂和使用该催化剂的氨合成法的制作方法
本发明涉及一种在载体上负载有金属的催化剂,特别是一种以电子化合物(electride)为载体的氨合成用的负载金属催化剂和使用该催化剂的氨合成法,所述电子化合物是电子在层状化合物的层间以阴离子电子存在的物质。
背景技术:
以M3N2(M为Ca、Sr或Ba)表示的碱土金属氮化物应用于半导体装置所用的氮化铝原料、金属滑动部件用的陶瓷粒子、电池电极构成材料、导电粒子等(专利文献1)。专利文献1公开了加热分解第二族金属氨基化合物制成M3N2(M为Be、Mg、Ca、Sr或Ba)或M2N(M为Be、Mg、Ca、Sr或Ba)等的方法。专利文献2公开了将氨与第二族金属反应,使其液化得到第二族金属氨基化合物,并将该第二族金属氨基化合物加热分解制成高纯度M3N2(M为Be、Mg、Ca、Sr或Ba)的方法。
至今为止,α-Ca3N2、β-Ca3N2、γ-Ca3N2、Ca11N8、Ca2N等作为氮化钙已为人所知。另外,氮化钙的氢化物Ca2NH、CaNH、Ca(NH2)2等(以下也称为“Ca-N-H系化合物”)也已为人所知。
Ca3N2是使Ca溶解在液氨中,并在氮气环境下加热分解而制得。如以下反应式所示,Ca3N2与空气中的水分反应生成氢氧化钙和氨。该反应在水中也可以进行。
Ca3N2+6H2O→3Ca(OH)2+2NH3
另外,已知Ca2N容易被氧化,是极不稳定的物质,已有报道称Ca2N稳定存在的范围是1000℃以下的氩气中或者250℃至800℃的氮气中(非专利文献1)。
另一方面,本发明人在2013年发现了Ca2N是二维电子化合物(非专利文献2)。Ca2N:e-是电子作为阴离子结合在由[Ca2N]+构成的层间而形成的层状化合物,可通过在真空条件下加热Ca3N2和金属Ca而制得。已有报道称Ca2N:e-的传导电子浓度为1.39×1022/cm3,功函数为2.6eV。然后,在非专利文献3中报道了该二维电子化合物。
本发明人还针对具有层状结晶结构并由以离子式[AE2N]+e-表示的氮化物(AE为选自Ca、Sr、Ba中的至少一种元素)组成的氮化物电子化合物相关的发明提出了专利申请(专利文献3)。
另一方面,以含有氢气和氮气的气体为原料,以氧化铁为催化剂,以氧化铝、氧化钾为促进剂的哈伯-博施(Haber-Bosch)氨合成法已得到普及。该方法100年来无大的变化。采用哈伯-博施法进行氨合成是在300℃~500℃、20~40MPa的高温高压下,使氮气和氢气在催化剂上进行反应而进行。
VIII族金属和VIB族金属的三元氮化物(专利文献4)、钴-钼络合氮化物(专利文献5)等作为氨合成用催化剂也已为人所知。本发明人开发了以钙铝石型化合物为氨合成催化剂载体的负载金属催化剂(专利文献6~7,非专利文献4~5)。
现有技术文献
专利文献
专利文献1:日本特许第4585043号公报(US2012225005A1)
专利文献2:日本特开2012-66991号公报(特许第5627105)
专利文献3:日本特开2014-24712号公报
专利文献4:日本特开2000-264625号公报(US6235676B1)
专利文献5:日本特开2001-096163号公报(专利第4038311)
专利文献6:WO2012/077658(US2013183224A1)
专利文献7:WO2014/034473
非专利文献
非专利文献1:P.Hchn,S.Hoffmann,J.Hunger,S.Leoni,F.Nitsche,W.Schnelle,R.Kniep,"Chem.Eur.J.",15,3419(2009)
非专利文献2:K.Lee,S.W.Kim,Y.Toda,S.Matsuishi and H.Hosono,"Nature",494,336-341(2013)
非专利文献3:A.Walsh and D.O.Scanlon,"Journal of materials Chemistry"C,1,3525-3528(2013)
非专利文献4:M.Kitano,Y.Inoue,Y.Yamazaki,F.Hayashi,S.Kanbara,S.Matsuishi,T.Yokoyama,S.W.Kim,M.Hara and H.Hosono,”Nature Chemistry”vol.4,934-940(2012)
非专利文献5:Y.Inoue,M.Kitano,S.W.Kim,T.Yokoyama,M.Hara and H.Hosono,”ACS Catal.”4(2),674-680(2014)
技术实现要素:
发明要解决的课题
Ca3N2应用于半导体装置所用的氮化铝原料、金属滑动部件用的陶瓷粒子、电池电极构成材料、导电粒子等,然而,已知Ca2N是极不稳定的物质,其用途目前基本上尚未开发,也尚未进行对其结构、物理性质等的研究。
氨合成的催化剂多数采用以Fe粒子为活性金属的催化剂,近年来也使用以活性炭为载体的Ru/C催化剂。发现氧化铁催化剂以来,已研究开发了各种催化剂金属、载体材料,几乎用尽了已知金属+载体的组合。目前很难找到一种活性远远超过由已知金属+载体的组合可实现的活性的催化剂,特别是,即使不使用大量促进剂也具有高活性且化学稳定、热稳定的新催化剂。
解决课题的方法
本发明人在研究氮化钙的物质结构、物理性质过程中,发现如上所述,层状化合物Ca2N是电子作为阴离子结合在由[Ca2N]+构成的层间的层状化合物,即,由层间存在的电子来结合层相互之间的电子化合物。该电子化合物以示构式[Ca2N]+·e-表示,由于在层状化合物的各层间隙,即二维间隙中存在根据化学计量预计浓度的电子,因此,可称为“二维电子化合物”。在二维电子化合物中,由于电子以二维非定域阴离子的电子形式存在于空隙中,因此可以在整个物质中极其顺畅地来回移动。
本发明人发现尽管Ca2N是极不稳定的物质,但是氮化钙的高温反应特性出人意料地对以含有氢气和氮气的气体为原料进行的连续氨合成极其有效。
即,发现如果将Ru、Fe、Co等对氨合成有催化活性的过渡金属(以下称为“Ru等过渡金属”)负载在Ca2N上而形成负载金属催化剂时,可飞跃性提高氨合成活性,并在未使用不稳定的碱金属、碱土金属及其化合物作为促进剂的情况下,可在长时间的合成反应中仍保持稳定,成为性能显著优于现有催化剂的氨合成催化剂。
另外,也了解到当将Ru等过渡金属催化剂负载在一种氮化钙Ca3N2上时,在氨合成反应过程中,载体结构由Ca3N2变为二维电子化合物的Ca2N结构,表现出高催化活性。即,Ca3N2可用作由Ca2N组成的二维电子化合物的前体。
如上所述,高催化活性是由二维电子化合物产生的特有的特性,将表示为与Ca2N相同的离子式[AE2N]+e-的氮化物Sr2N、Ba2N作为载体,也能够得到相同的效果。另外,Ca2N的Ca,Sr2N的Sr,Ba2N的Ba的一部分可替换为选自于由Li、Na、K、Rb、Cs所组成的组中的至少一种以上的碱金属元素。
另外,认为:如上所述将由与Ca2N相同的层状化合物Y2C、Sc2C、Gd2C、Tb2C、Dy2C、Ho2C、Er2C等碳化物组成的二维电子化合物及其前体物质作为载体时,高催化活性是共同的普遍特性。本发明人已证实:如实施例5所示,以碳化物Y2C作为Ru等过渡金属的载体而形成的催化剂,也具有与以Ca2N作为载体时同样高的催化活性。Y2C可与Ca2N相同地形式上表示为离子式[Y2C]2+·2e-,另外,认为反应过程中相间存在的一部分电子很容易置换为氢而形成氢化物结构,因此推断这些氢化物也有利于催化活性。
另外,氮化钙的氢化物Ca2NH、CaNH、Ca(NH2)2等(以下也称为“Ca-N-H系化合物”)已为人所知,这些氢化物作为Ca2N的前体,或者,在氨合成反应过程中Ca3N2在反应中变为Ca2N时,形成Ca2NH、CaNH这类氢化物的结构,推测这些化合物也有利于催化活性。
以前存在计算将Sr3N2、Be3N2、Ca3N2、Ba3N2、Mg3N2和氢反应生成氨的反应的文献(非专利文献4),但是这些反应是化学计量反应,不能连续生成氨。另一方面,采用本发明的催化剂的方法,通过将过渡金属纳米粒子负载在二维电子化合物上形成催化剂,可连续生成氨。
非专利文献4:Thermodynamics of synthesis of ammonia through a calcium nitride-hydride system,Kondrat'ev,A.V.;Beglov,B.M.;Rylov,A.A.Deposited Doc.Avail.BLLD.(1975),(VINITI 2626-75),10pp
本发明的负载金属催化剂可采用浸渍法、物理混合法、热分解法、液相法、蒸镀法等方法制成。优选地,利用Ru等过渡金属络合物的加热分解,在构成载体的化合物粉末或成型体上蒸镀Ru等过渡金属,从而进行负载并制成负载金属催化剂。在氨合成反应装置中使用所述催化剂,在100℃以上且600℃以下的反应温度,优选在10kPa以上且低于20MPa的反应压力条件下,使原料氮气和氢气在催化剂上反应,从而可连续高效地合成氨。
例如,将Ru负载在如Ca2N、Y2C等二维电子化合物上时,本发明的催化剂的活性高于目前所用催化剂中活性水平最高的负载金属催化剂Ru-Cs/MgO的活性,表现出与Ru/C12A7:e-同等以上的活性,所述Ru/C12A7:e-是本发明人以前开发的以含有1018cm-3以上传导电子的钙铝石型化合物为载体的负载金属催化剂。另外,将Ru负载到Ca3N2上时,在氨反应过程中载体结构由Ca3N2变为二维电子化合物Ca2N和Ca2NH、CaNH、Ca(NH2)2等“Ca-N-H系化合物”,在340℃以下的低温区域中也表现出比Ru/C12A7:e-高2倍以上的催化活性。
发明效果
在采用本发明的催化剂通过氢气和氮气反应来连续合成氨的方法中,采用仅由钙、氮、氢等克拉克数(クラーク数)排位靠前的元素构成的廉价化合物,而且,优选地,在10kPa以上且低于20MPa的压力,更优选地在10kPa~10MPa的低反应压力条件下,消耗少量能量即可合成氨,而且,即使重复进行合成反应仍未发现催化活性降低,从而能够以长期化学稳定性、热稳定性,高效地合成氨。
另外,也可以使用除Ru等高价稀有金属以外的过渡金属元素,例如,Fe、Co等作为负载物,从有效利用资源的角度看也是有用的。另外,也可不需要像现有氧化铝等载体那样添加碱金属、碱金属化合物、碱土金属化合物等促进化合物,因此制造简单。
附图说明
图1是表示使用在各种载体上负载Ru的催化剂进行氨合成反应的催化活性的图示。
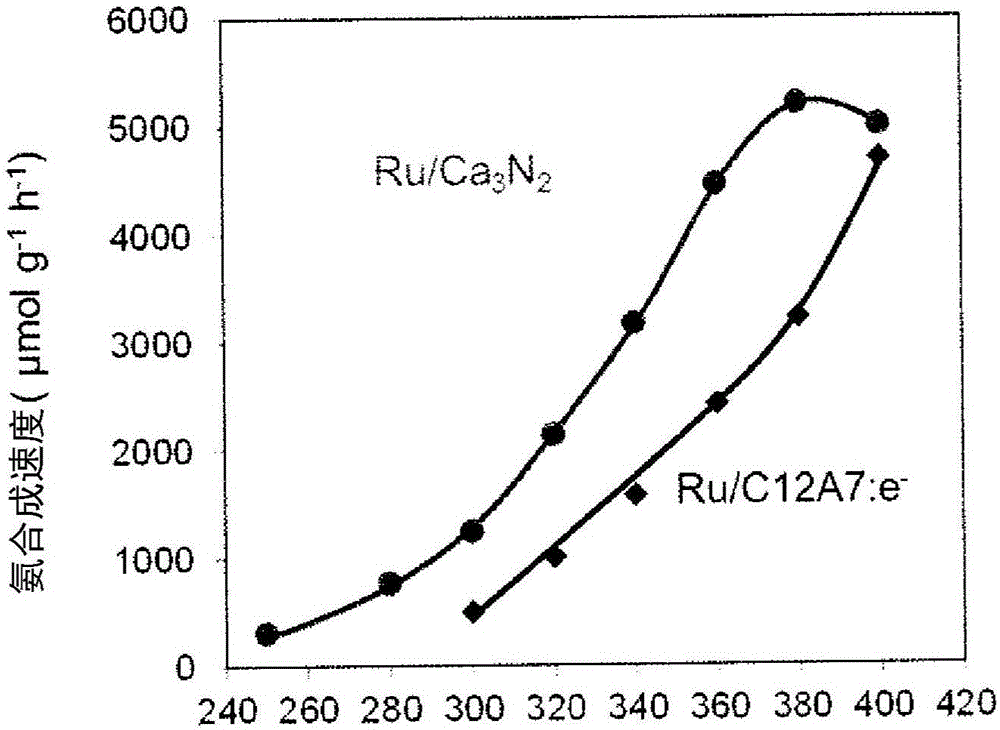
图2是表示使用Ru负载Ca3N2和Ru负载C12A7:e-在各种反应温度(横轴:℃)进行氨合成反应时的氨合成速度(纵轴)的图示。
图3是表示在与氨合成反应相同的条件下Ru负载Ca3N2(a)和Ca2N(b)的拉曼光谱结果的图。
图4是表示对实施例2的催化剂进行稳定性评价试验的结果的图示(横轴:反应时间,纵轴:氨合成速度)。
具体实施方式
本发明涉及以含有氢气和氮气的气体为原料进行连续氨合成所用的催化剂。在所述催化剂中,负载具有催化活性的过渡金属的载体是二维电子化合物或其前体。
所述二维电子化合物或其前体以MxNyHz(其中,M为选自于由Mg、Ca、Sr和Ba所组成的组中的一种或两种以上第二族金属,x、y、z的范围为1≤x≤11、1≤y≤8、0≤z≤4,x为整数,y、z不限于整数)表示。在该式中,当z=0时,为金属氮化物,0<z时为氢化物。N有缺失的情况,y不限于整数。另外,由于氢被引入金属氮化物中,因此,z不限于整数。
典型的金属氮化物可以举出,当x=3、y=2、Z=0时,以M3N2(其中,M为选自于由Mg、Ca、Sr和Ba所组成的组中的一种或两种以上第二族金属)表示的金属氮化物。具体的金属氮化物可以举出,当M为Ca时,以Ca3N2或Ca2N表示的钙氮化物。所述二维电子化合物或其前体不限于金属氮化物,也可以是选自于由Y2C、Sc2C、Gd2C、Tb2C、Dy2C、Ho2C和Er2C所组成的组中的金属碳化物或其氢化物。
下面以氮化钙为具体例子详细说明本发明的催化剂和该催化剂的制造方法,以及使用该催化剂的氨合成方法(下面称为“本发明的方法”),对其他二维电子化合物可同样实施。
作为钙氮化物系化合物(包括氢化物),α-Ca3N2、β-Ca3N2、γ-Ca3N2、Ca11N8、Ca2N、Ca2NH、CaNH、Ca(NH2)2等已为人所知,Ca最大可为11,N最大可为8,H最大可为4。因此,本发明的催化剂中的钙氮化物和“Ca-N-H系化合物”,总括地以通式CaxNyHz(其中,M为选自于由Mg、Ca、Sr和Ba所组成的组中的一种或两种以上第二族金属,x、y、z的范围为1≤x≤11、1≤y≤8、0≤z≤4,x为整数,y、z不限于整数)表示。0<z表示氢化物,当其为氢化物时,z可为小于1的数,不限于整数。
另外,考虑到在氨合成反应中除氮化钙之外的Mg3N2、Sr2N、Ba2N各金属氮化物也进行与Ca3N2相同的结构变化,因此,在本发明的催化剂中的氮化物或含有氢的氮化物可以用通式MxNyHz(其中,M为选自于由Mg、Ca、Sr和Ba所组成的组中的一种或两种以上第二族金属,x、y、z的范围为1≤x≤11、1≤y≤8、0≤z≤4,x为整数,y、z不限于整数)表示。
<氮化钙的合成>
氮化钙(Ca3N2)在市场中有售,也可以采用使金属钙溶解在液氨中,将得到的钙氨基化物在氮气流中加热分解的方法等制得。例如,将Ca3N2和金属钙一同在真空中加热至1000℃左右可得到氮化钙(Ca2N)。
<Ca-N-H系化合物催化剂的合成>
以式CaxNyHz(x、y、z的范围为1≤x≤11、1≤y≤8、0<z≤4,x为整数,y、z不限于整数)表示的氮化钙的氢化物用作载体也表现出与Ca2N相同的高催化活性。举出Ca2NH、CaNH、Ca(NH2)2等作为这些Ca-N-H系化合物的具体例子。
将Ru等过渡金属元素催化剂负载在氮化钙(Ca3N2)上进行氨合成,在合成反应过程中,Ca3N2被还原,变成Ru负载Ca2N、Ru负载Ca-N-H系化合物或者这些物质的混合物。另外,作为其他方法,将Ca3N2或Ca2N在氢气流中加热至500℃左右,可得到Ca-N-H化合物,将Ru等过渡金属元素负载在该Ca-N-H化合物上,也具有催化剂的功能。
<Ru等过渡金属的负载工序>
已知过渡金属元素在各种合成反应中用作均相、非均相催化剂,特别是,Fe、Ru、Os、Co、Mo等6族、8族或9族过渡金属适于用作氢气和氮气直接反应合成氨的催化剂。在本发明中,可将选自于Cr、Mo、W中的6族金属,选自于Mn、Tc、Re中的7族,选自于Fe、Ru、Os中的8族,选自于Co、Rh、Ir中的9族的各元素单独或组合用作过渡金属元素。另外,也可使用这些元素的化合物,例如,Co3Mo3N、Fe3Mo3N、Ni2Mo3N、Mo2N。
使用氮化钙、Ca-N-H系化合物用作载体时,将所述通过氮化钙合成工序制得的物质与Ru等过渡金属化合物采用浸渍法、物理混合法混合。例如,物理混合法可以举出:将Ru等金属络合物和氮化钙在研钵等中一边粉碎一边混合的方法;封装至玻璃管后通过振摇或旋转混合的方法;采用球磨机粉碎混合的方法等。
使用固体烧结体、薄膜、固体单晶等作为载体时,可与粉末、多孔材料同样地采用浸渍法,采用化学蒸镀法(CVD法)、溅射法等使Ru等过渡金属化合物在其表面堆积,加热分解该过渡金属化合物,使过渡金属析出等方法。当使用所述过渡金属化合物时,例如,可采用通过CVD法等使各金属原料在载体上堆积,加热分解后用氨气氮化的方法。
所述过渡金属化合物无特别限定,例如,可列举出十二羰基三钌[Ru3(CO)12]、二氯四(三苯基膦)钌(II)[RuCl2(PPh3)4]、二氯三(三苯基膦)钌(II)[RuCl2(PPh3)3]、三(乙酰丙酮)钌(III)[Ru(acac)3]、二茂钌[Ru(C5H5)]、氯化钌[RuCl3]、五羰基铁[Fe(CO)5]、碘化四羰基铁[Fe(CO)4I2)]、氯化铁[FeCl3]、二茂铁[Fe(C5H5)2]、三(乙酰丙酮)铁(III)[Fe(acac)3]、十二羰基三铁[Fe3(CO)12]、氯化钴[CoCl3]、三(乙酰丙酮)钴(III)[Co(acac)3]、乙酰丙酮钴(II)[Co(acac)2]、八羰基钴[Co2(CO)8]、二茂钴[Co(C5H5)2]、十二羰基三锇[Os3(CO)12]、六羰基钼[Mo(CO)6]等加热易分解的无机金属化合物或有机金属络合物。
浸渍法可采用以下工序。例如,将载体粉末或成型体分散在过渡金属化合物溶液(例如,Ru羰基络合物的正己烷溶液)中,搅拌。此时,过渡金属化合物占载体粉末的0.01~40wt%、优选为0.02~30wt%、更优选为0.05~20wt%左右。然后,在氮气、氩气、氦气等非活性气体流或真空中,在50~200℃温度加热30分钟至5小时,蒸发溶剂,干燥。然后,还原由干燥的过渡金属化合物组成的催化剂前体。经以上工序将直径为数nm~数百nm的Ru等过渡金属微粒负载在载体粉末上,制得负载金属催化剂。
所述负载金属催化剂的比表面积为0.1~200m2/g左右,Ru等过渡金属的负载量相对于载体粉末或成型体为0.01~30wt%,优选为0.02~20wt%、更优选为0.05~10wt%。
另外,替代上述方法,在相同条件下通过物理混合法将氮化钙、Ca-N-H系化合物与Ru等过渡金属化合物粉末固相混合后加热,将Ru等过渡金属化合物还原分解为Ru等过渡金属,从而可得到同样形式的负载金属催化剂。
另外,本发明的负载金属催化剂可使用常用的成型技术以成型体的形式使用。具体而言,可以举出粒子形、球形、片形、环形、管形、四叶形、六面体、蜂窝体等形状的成型体。另外,也可将本发明的负载金属催化剂在合适的支撑体上涂层后使用。
<氨合成>
本发明的氨合成方法是使用所述负载金属催化剂作为催化剂,使氮气和氢气在催化剂上反应来合成氨的方法。反应的一个典型实施方式是与现有的哈伯-博施法相同,使氮气和氢气混合气体原料在加热加压条件下直接反应,将基于N2+3H2→2NH3反应生成的氨冷却或用水吸收、分离的方法。供给氮气和氢气使其接触反应器内放置的负载金属催化剂。除去生成的氨后,回收循环反应器内未反应的氮气和氢气。供给氮气和氢气前,用氢气或氮气和氢气混合气体还原处理负载金属催化剂的表面,优选采用前处理还原除去负载的Ru等过渡金属上附着的氧化物等。
氮化钙、Ca-N-H系化合物在大气中优先吸附水分,另外,由于在过量水分存在下化合物自身会发生分解,因此,氨合成反应尽可能在不含有水分的条件下进行,即,优选的是使用含水量为100ppm以下,优选为50ppm以下的氮气和氢气为原料。
然后,在原料氮气和氢气混合气体环境下,加热负载金属催化剂以合成氨。优选在氮气和氢气的摩尔比约为1/10~1/1,优选为1/5~1/1的条件下进行。反应温度优选为100℃以上且低于600℃,优选为200℃以上至500℃左右,更优选为250℃以上至500℃左右。反应温度越低越有利于平衡向氨生成方向进行,为了在得到足够的氨生成速度的同时,有利于平衡向氨生成方向进行,优选采用上述范围。
进行合成反应时氮气和氢气混合气体的反应压力无特定限制,优选为10kPa以上且低于20MPa,考虑到实际应用,优选在加压条件下进行反应,实际应用中,更优选为100kPa~20MPa左右,在低于3MPa的条件下也很有可能进行合成反应。
合成反应方式可以是间歇式反应方式、封闭循环式反应方式、流通式反应方式中的任一种,从实际应用的角度看最优选流通式反应方式。氨合成反应是平衡关系,在高压且低温条件下有利于反应进行。另外,由于是放热反应,除去反应热有利于反应进行,采用各种方法以提高工业上的收率。例如,当采用流通式反应装置时,建议串联多个填充了催化剂的反应器,在各反应器出口安装中间冷却器进行散热,从而降低各反应器的入口温度,得到较高的氨收率的方法。另外,建议使用内部具有多个填充了Fe催化剂和Ru系催化剂的催化剂层的反应器,精确控制各反应层的出口温度的方法。
在本发明中,与现有方法相同,可使用填充催化剂的一种反应器或多个反应器进行氨合成反应。使用的催化剂可以是本发明的单个催化剂或选自本发明的催化剂中的两种以上催化剂的组合、或本发明的催化剂和公知催化剂的组合中的任一种。另外,可以使用连结多个反应器的方法、同一个反应器内设置多个反应层的方法等中的任何方法。
在本发明中,当组合使用催化剂时,由于本发明的催化剂在低温下活性高,因此优选在最后的反应器中使用。即,通过在对平衡有利的低温下进行最终反应,能够得到高的氨收率。
在工业氨合成的平衡反应条件下,为了限制平衡,反应器出口的反应气体中氨浓度为20%以下,从而冷却并除去反应气体中的生成氨后,将反应气体、未反应原料中含有的部分杂质通过膜分离等方法分离,并清除到体系外后,回收使用未反应原料作为原料。
氨合成法的氢气原料可以是使用煤炭、石油或天然气为原料,采用水蒸气改质法、部分氧化改质法、或结合水蒸气改质和部分氧化的自热改质法、变换反应相结合制造的方法、使用生物质为原料的方法、电解水的方法、光催化剂分解水的方法等各种方法制得的任意氢气原料。
当使用天然气原料作为氨合成法的原料时,氢气和氮气经由天然气的水蒸气改质工序和部分氧化改质工序、CO变换反应工序、除CO2工序、随后通过CO甲烷化除CO的工序而制成。由于所述水蒸气改质反应是吸热反应,因此可利用自热反应产生的反应热,使用空气作为氮气原料时,H/N摩尔比为1.7-2.5摩尔比左右。由于水蒸气改质的未反应气体含有氢气,因此优选在改质工序中将其作为回收气体循环使用,开发了通过控制新鲜气体和回收气体的比例有效进行反应的方法,在本发明中,同样可使用前述方法。
另一方面,开发了使用富氧空气制得高H/N比原料的方法,由于使用该原料可使回收气体量减少,因此该方法是从能源的角度优选的方法。另外,压缩分离空气后,氧气可用于自热法制氢,氮气可用于反应气体、工业氮气等,从节能角度看该方法是优选的方法,在本发明中可使用任一原料制造方法。
下面,基于实施例更详细地说明本发明。采用气相色谱仪求得NH3的生成量,或将生成的NH3溶解在硫酸水溶液中,采用离子色谱仪定量该溶液,求得氨合成速度,从而评价本发明和比较例的催化剂的氨合成活性。
实施例1
<负载Ru的Ca3N2的制备>
将1g市售试剂Ca3N2粉末(BET表面积约为1m2g-1)在Ar环境的手套箱内与0.042g的Ru3(CO)12粉末混合,并装入真空的石英玻璃管内。在转动装入玻璃管的样品的同时,在250℃温度下加热15小时。由此制得负载2wt%的Ru金属粒子的Ca3N2催化剂。所述催化剂的BET表面积约为1m2g-1,负载后BET表面积几乎无变化。采用CO吸附法测定Ru分散度(%)为3.0。
<氨合成反应>
进行氮气(N2)和氢气(H2)反应生成氨(NH3)的合成反应。在反应温度为400℃时,将0.2g上述方法制得的催化剂装入玻璃管,在反应温度为340℃时,将0.1g上述催化剂装入玻璃管,采用固定床流通式反应装置进行合成反应。在气体流量为N2:15mL/min,H2:45mL/min,合计60mL/min,压力为大气压的条件下进行反应。将从流通式反应器出来的气体通入0.005M硫酸水溶液中,使生成的氨溶解在溶液中,采用离子色谱仪对生成的铵离子进行定量。在400℃条件下氨合成速度如图1所示,为2760μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为3164μmolg-1h-1。TOF(×10-3s-1)为164.2。
实施例2
除使用Ca2N(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Ca2N催化剂,在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为2750μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为3386μmolg-1h-1。TOF(×10-3s-1)为171.2。
实施例3
除使用Ca(NH2)2(BET表面積为120m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Ca(NH2)2催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表1所示,为2118μmolg-1h-1。TOF(×10-3s-1)为3.2。
实施例4
除使用CaNH(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/CaNH催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表1所示,为1107μmolg-1h-1。TOF(×10-3s-1)为38.4。
实施例5
除使用Y2C(BET表面積为0.2m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Y2C催化剂,在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为580μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为1101μmolg-1h-1。TOF(×10-3s-1)为59.4。
表1示出了使用在各种载体上负载Ru制得的催化剂进行氨合成反应的结果(反应温度:340℃,流量:60mLmin-1,压力:0.1MPa,N2:H2=1:3)。
表1
实施例6
除使实施例1的Ca3N2负载的Ru量为0.5wt%之外,采用与实施例1相同的方法制备0.5wt%Ru/Ca3N2催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表2所示,为2009μmolg-1h-1。
实施例7
除使实施例1的Ca3N2负载的Ru量为1.0wt%之外,采用与实施例1相同的方法制备1.0wt%Ru/Ca3N2催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表2所示,为2399μmolg-1h-1。
实施例8
除使实施例1的Ca3N2负载的Ru量为3.0wt%之外,采用与实施例1相同的方法制备3.0wt%Ru/Ca3N2催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表2所示,为3446μmolg-1h-1。
实施例9
除使实施例1的Ca3N2负载的Ru量为5.0wt%之外,采用与实施例1相同的方法制备5.0wt%Ru/Ca3N2催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表2所示,为3922μmolg-1h-1。
表2示出了使用Ru负载量不同的Ca3N2进行的氨合成反应的结果(反应温度:340℃,流量:60mLmin-1,压力:0.1MPa,N2:H2=1:3)。
表2
实施例10
<负载Co的Ca3N2的合成>
将1g市售试剂Ca3N2粉末在Ar环境的手套箱内与0.058g的Co2(CO)8粉末混合,并装入真空的石英玻璃管内。将装入玻璃管的样品在250℃条件下加热15小时。由此制得负载2wt%Co金属的Ca3N2催化剂。该催化剂的BET表面积约为1m2g-1。使用合成的催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表3所示,为439μmolg-1h-1。
实施例11
<负载Fe的Ca3N2的合成>
将1g市售试剂Ca3N2粉末在Ar环境的手套箱内与0.065g的Fe2(CO)8粉末混合,并装入真空的石英玻璃管内。将装入玻璃管的样品在250℃条件下加热15小时。由此制得负载2wt%Fe金属的Ca3N2催化剂。该催化剂的BET表面积约为1m2g-1。使用合成的催化剂,在与实施例1相同的条件下实施氨合成反应。在340℃条件下氨合成速度如表3所示,为284μmolg-1h-1。
表3示出了使用负载各种过渡金属的Ca3N2进行的氨合成反应的结果(反应温度:340℃,流量:60mLmin-1,压力:0.1MPa,N2:H2=1:3)。
表3
实施例12
除使用Mg3N2(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Mg3N2催化剂,在实施例1的条件下实施氨合成反应。在340℃条件下氨合成速度如表4所示,为365μmolg-1h-1。TOF(×10-3s-1)为16.9。
实施例13
除使用Sr2N(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Sr2N催化剂,在实施例1的条件下实施氨合成反应。在340℃条件下氨合成速度如表4所示,为1520μmolg-1h-1。TOF(×10-3s-1)为70.2。
实施例14
除使用Ba2N(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Ba2N催化剂,在实施例1的条件下实施氨合成反应。在340℃条件下氨合成速度如表4所示,为566μmolg-1h-1。TOF(×10-3s-1)为26.1。
表4示出了使用在各种金属氮化物载体上负载Ru(2wt%)的催化剂进行氨合成反应的结果(反应温度:340℃,流量:60mLmin-1,压力:0.1MPa,N2:H2=1:3)。
表4
[比较例1]
<钙铝石型化合物粉末的合成>
将CaCO3粉末和Al2O3粉末以Ca:Al比为11:14的比例混合,将总计30g置于氧化铝坩埚内,在1300℃条件下加热6小时。将得到的粉末装入石英玻璃管内,在1×10-4Pa的真空中,在1100℃加热15小时,得到钙铝石型化合物粉末原料。该阶段的比表面积为1m2g-1以下。
<通过还原处理的电子导入>
将3g由此得到的粉末和0.18g金属Ca粉末一同装入石英玻璃管,在700℃加热15小时,从而使内部以金属Ca蒸汽环境与粉末反应。取出封入真空管内的样品,用研钵研碎后再装入石英玻璃管内,在抽真空的同时封管。将其在1100℃加热2小时,从而得到传导电子浓度约为2×1021cm-3、比表面积为1m2g-1的导电性钙铝石型化合物粉末C12A7:e-。除使用C12A7:e-之外,在与实施例1相同的条件下负载2wt%Ru,实施氨合成反应。在400℃条件下氨合成速度如图1所示,为2684μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为1571μmolg-1h-1。TOF(×10-3s-1)为52.1。
[比较例2]
除使用不含传导电子的化学当量组成的C12A7(非掺杂型)代替比较例1的导电性钙铝石型化合物之外,采用与实施例1相同的方法制备2wt%Ru/C12A7催化剂,在实施例1的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为546μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为88μmolg-1h-1。TOF(×10-3s-1)为2.7。
[比较例3]
除使用CaO·Al2O3(表示为CA)(BET表面積为1m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/CA催化剂,在实施例1的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为467μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为22μmolg-1h-1。TOF(×10-3s-1)为0.7。
[比较例4]
除使用添加了Cs的MgO(表示为Cs/MgO)(BET表面積为12m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru-Cs/MgO催化剂(Cs/Ru元素比=1),在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为2264μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为2367μmolg-1h-1。TOF(×10-3s-1)为13.3。
[比较例5]
除使用添加了Ba的活性炭(表示为Ba/AC)(BET表面積为310m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru-Ba/C催化剂(Ba/Ru元素比=1),在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为148μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为29μmolg-1h-1。TOF(×10-3s-1)为0.2。
[比较例6]
除使用CaO(BET表面積为3m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/CaO催化剂,在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为158μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为261μmolg-1h-1。TOF(×10-3s-1)为7.5。
[比较例7]
除使用Al2O3(BET表面積为170m2g-1)代替实施例1的Ca3N2之外,采用与实施例1相同的方法制备2wt%Ru/Al2O3催化剂,在与实施例1相同的条件下实施氨合成反应。在400℃条件下氨合成速度如图1所示,为8.5μmolg-1h-1。在340℃条件下氨合成速度如表1所示,为10μmolg-1h-1。TOF(×10-3s-1)为0.1。
[氨合成速度的比较]
从表1所示的氨合成速度来看,可知与在现有催化剂载体(Al2O3、CaO、Ba/AC、Cs/MgO、CA)上负载Ru的催化剂相比,尽管负载Ru的Ca3N2、Ca2N比表面积小,然而在单位重量相同的情况下,其表现出高催化活性、高TOF值,是非常优越的催化剂。可知该催化活性表现出比负载Ru的C12A7:e-更高的值。
另一方面,实施例5中负载Ru的Y2C每单位重量的氨合成速度比Ru/C12A7:e-差,但是,考虑到其BET表面积为0.2m2/g-1,因此其单位表面积的催化活性与Ru/C12A7:e-相当。
[评价试验1]
除反应温度以外,在与实施例1相同的条件下进行氨合成反应,并评价催化剂的反应温度依赖性。在催化剂用量为0.1g,气体流量为N2:15mL/min,H2:45mL/min,总计60mL/min,压力为大气压的条件下进行反应。图2表示了使用2wt%Ru/Ca3N2和2wt%Ru/C12A7:e-作为催化剂在各种反应温度下进行氨合成反应的结果。在400℃条件下二者活性几乎相同,而在340℃以下的低温度区域内,显然2wt%Ru/Ca3N2的催化活性比2wt%Ru/C12A7:e-高2倍左右。
[评价试验2]
图3表示了除反应温度之外,在与实施例1相同的条件下进行氨合成反应的同时,采用拉曼光谱检测2wt%Ru/Ca3N2催化剂(a)和2wt%Ru/Ca2N催化剂(b)的结构变化的结果。升高反应温度时,2wt%Ru/Ca3N2在258cm-1出现的Ca-N伸缩振动产生的峰减少,反应温度达到350℃时,观察到在180cm-1和322cm-1有两个峰。
另一方面,在2wt%Ru/Ca2N也在氨合成条件下的氮气和氢气流中加热时,观察到在180cm-1和322cm-1有两个峰。从这些结果来看,显然在氨合成反应条件下,在350℃加热时,2wt%Ru/Ca3N2变为与Ca2N类似的结构,表现出高催化活性。
[评价试验3]
在340℃的反应温度下,继续进行20小时的合成反应,并评价催化剂的稳定性。图4表示了使用实施例2的2wt%Ru/Ca2N作为催化剂,进行氨合成的结果。可知在反应了约20小时时,所述催化剂仍保持稳定,可生成氨,反应活性几乎没有降低。
表2表示了除Ru之外,将Co、Fe负载在Ca3N2上的催化剂在反应温度340℃的氨合成活性。与实施例1的2wt%Ru/Ca3N2相比,负载Co、Fe的催化剂(实施例6、实施例7)的性能较差,但是仍表现出与除比较例4的Ru-Cs/MgO以外的现有Ru催化剂相当或更好的催化性能。
在钙氮化物(Ca3N2、Ca2N)和镁氮化物、锶氮化物、钡氮化物(Mg3N2、Sr2N、Ba2N)上负载2wt%Ru的催化剂在反应温度340℃的氨合成活性如表3所示。实施例8的2wt%Ru/Mg3N2、实施例9的2wt%Ru/Sr2N、实施例10的2wt%Ru/Ba2N催化剂与2wt%Ru/Ca3N2、2wt%Ru/Ca2N相比,催化性能较差,但是仍表现出比除比较例4的Ru-Cs/MgO以外的现有Ru催化剂更高的催化性能。
工业实用性
目前,在生产氨时通常采用Fe3O4中含有一定重量%的Al2O3和K2O的双重促进铁催化剂的合成方法(哈伯-博施法),在哈伯-博施法中,需要约20MPa以上的高压,而本发明的方法不需要高压,在低于20MPa,甚至是低于3MPa的较低压下也可以充分进行合成反应,可以简化生产过程、降低能量消耗,因此是优选的方法。另外,比现有的Ru催化剂相比价格更便宜,能够非常高效地生产氨。
- 还没有人留言评论。精彩留言会获得点赞!