MoS2/CoS2复合裂解水产氢低过电位电催化剂及其硫化法制备方法与流程
本发明属于催化剂制备技术领域,具体涉及一种MoS2/CoS2复合裂解水产氢低过电位电催化剂及其硫化法制备方法。
背景技术:
随着全球工业化进程的加快,煤、石油、天然气这三大工业生产赖以生存的支柱能源,将面临被耗尽枯竭的局面,氢能作为一种新型能源,具有热值高,无污染,原料来源广泛等天然优势,受到世界各国越来越多的关注,那么如何制备可持续的氢能源是全世界能源科学家共同关心的问题,电催化裂解水产氢是一种能可持续、大规模产生氢气的一种手段,但是电催化水产氢面临的最大问题是氢气发生所需要的过电位过高,带来能耗的增加、成本高,严重限制了氢能源的推广应用,目前,贵金属基催化剂(比如Ru,Pt或Ir的氧化物)是电催化性能最好的水裂解催化剂。但是,价格昂贵、地球储量低的缺点极大地限制了该类催化剂的广泛应用,因此开发高效、低过电位的非贵金属电催化剂就显得尤为紧迫。
技术实现要素:
为了解决现有技术中非贵金属电催化剂存在的过电位过高对能源浪费严重的问题,本发明对电催化裂解水产氢的机理进行了深入研究,在付出了大量的原创性劳动后,进而完成了本发明。
本发明所述的电催化剂具体是由硝酸钴、氯化钴、乙酸钴、硫酸钴中的一种和钼酸钠、钼酸铵中的一种采用水热法制备前驱体,前驱体再经过水洗去除未反应的钠离子、钴离子等杂质后干燥,然后再将一定量的干燥的前驱体和适量的升华硫分别放在两个石英舟里,在氮气保护的环境中通过控制一定的升温速率和焙烧温度,焙烧一定时间后即得到本发明所述的MoS2/CoS2复合裂解水产氢低过电位电催化剂。这种电催化剂在交流电流密度-60mA/cm-2时的过电位仅为-287mV,这是在非贵金属电催化剂中具有较低过电位的电催化剂。相比单纯的MoS2在过电位-287mV时交流电流密度仅为-3.68mA/cm-2提高了超过15倍;相比单纯的CoS2在过电位-287mV时交流电流密度仅为-43.36mA/cm-2提高了超过38.3%。因此本发明的电催化剂是一种具有可持续生产氢能源的一种先进技术手段之一,具有很好的工业应用前景,用以缓解当今社会严重依赖化石能源的局面。
本发明所述的一种MoS2/CoS2复合裂解水产氢低过电位电催化剂的硫化法制备方法,其步骤如下:
(1)将0.1~0.3克硝酸钴、氯化钴、乙酸钴或硫酸钴固体颗粒加入10~30毫升去离子水搅拌5~10分钟,使其溶解完全;
(2)将0.1~0.3克钼酸钠或钼酸铵固体颗粒加入10~30毫升去离子水搅拌5~10分钟,使其溶解完全;
(3)将步骤(1)和步骤(2)得到的溶液混合在一起,然后搅拌1~2小时;
(4)将步骤(3)得到的溶液转移到带有不锈钢衬底的反应釜中,再将该反应釜放入100~200摄氏度的烘箱中保持10~20小时,等反应釜温度降到室温时将反应釜中的上清液倒掉,底物依次用去离子水清洗3~6次,无水乙醇清洗2~4次,然后将洗净的产物放入60~120摄氏度的烘箱中干燥12~24小时,即得到所要制备前驱体;
(5)将步骤(4)制备的前驱体放入到石英舟内,将质量30~50倍于前驱体的升华硫放入另一石英舟内,然后将盛有前驱体的石英舟先推进管式炉加热区的石英管内,再将盛有升华硫的石英舟推入同一个管式炉加热区的石英管内,并保证盛有升华硫的石英舟位置在盛有前驱体的石英舟的后面,使得高温升华后的硫蒸气在氮气的引流下经过前驱体,从而使前驱体进行硫化;
(6)将步骤(5)管式炉加热区石英管内的空气排出,然后通入氮气,以30~50度/分钟的升温速率升温至400~450摄氏度,保温1.5~3小时进行焙烧硫化;当温度降到室温后取出盛有前驱体的石英舟,将所得产物浸泡在去离子水中超声处理20~40分钟,然后再依次用去离子水清洗离心3~6遍、无水乙醇清洗离心2~4遍,最后将所得产物放入80~120摄氏度的烘箱中干燥12~24小时,即得到本发明所述的MoS2/CoS2复合裂解水产氢低过电位电催化剂。
经法国生产的BioLogic电化学工作站的测试,本发明制备的电催化剂在0.5摩尔的硫酸溶液中在60mA/cm-2时的过电位仅为-287mV,相比单纯的MoS2在过电位-287mV时交流电流密度仅为-3.68mA/cm-2提高了超过15倍;相比单纯的CoS2在过电位-287mV时交流电流密度仅为-43.36mA/cm-2提高了超过38.3%。本发明可以使电催化产氢能耗得到降低,将进一步满足工业化推广的要求,为可持续生产氢能源提供一种有效的手段,因此本发明是一项十分有意义的发明创造。
附图说明
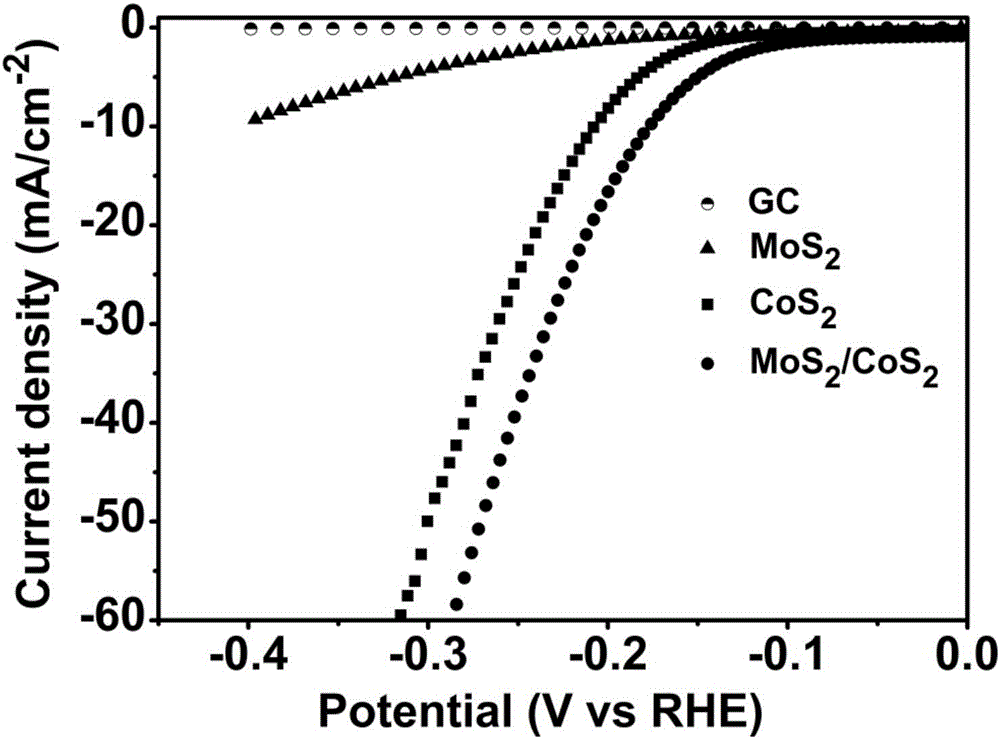
图1:纯GC(玻碳电极,实施例4制备)、MoS2、CoS2和本发明制备的MoS2/CoS2复合样品的交换电流密度和产氢过电位产氢(j vs V)表征图;从图中可以清楚的看出MoS2/CoS2复合样品的过电位最接近于0,接下来是CoS2的,再者是MoS2的,GC过电位与0的距离最远说明需要施加更高的负电压才能提高交换电流密度,从而可以看出MoS2/CoS2复合样品施加较低的负过电位就可以产生较大绝对值的交换电流密度。
图2:是MoS2/CoS2复合样品、CoS2和MoS2隧道扫描电子显微镜图(SEM);图中A、B、C是分别放大倍数为3万、8万、12万倍的MoS2/CoS2复合样品的扫描电镜照片;从这些电镜照片中可以看出本方法制备的产物的形貌是具有一些孔状的纳米材料。
图3:是CoS2样品的隧道扫描电子显微镜图(SEM);图中D、E、F是分别放大倍数为3万、8万、12万倍的CoS2的扫描电镜照片,从这些电镜照片中可以看出本方法制备的产物的形貌为生长的较为均一的纳米球。
图4:是样品MoS2隧道扫描电子显微镜图(SEM);图中F、H、I是分别放大倍数为3万、4.5万、7万倍的MoS2的扫描电镜照片,从这些电镜照片中可以看出本方法制备的产物的形貌十分漂亮的三维纳米花结构。
图5:对应实施例1产物的X射线衍射仪(XRD)的表征图,证明制备出了所要的MoS2/CoS2复合物,PDF卡片编号:37-1492和41-1471。
图6:对应实施例2产物的X射线衍射仪(XRD)的表征图,证明同样可以制备出了所要的CoS2,PDF卡片编号:41-1471。
图7:对应实施例3产物的X射线衍射仪(XRD)的表征图,证明制备出的产物MoS2,PDF卡片编号:37-1492。
具体实施方式
下面通过具体的实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
实施例1
(1)取0.291克六水合硝酸钴固体颗粒于反应容器中加入15毫升去离子水搅拌10分钟使其溶解完全;
(2)取0.242克二水合钼酸钠固体颗粒于另一反应容器中,再加入15毫升的去离子水搅拌10分钟至二水合钼酸钠完全溶解;
(3)将步骤(1)和步骤(2)中的溶液混合在一起,然后搅拌1.5小时;
(4)将步骤(3)中的溶液转移到带不锈钢衬底的100毫升的反应釜中,再将该反应釜放入已提前升温到140摄氏度的电热鼓风的烘箱中保持12小时,然后等反应釜温度降到室温时将反应釜中的上清液倒掉,底物采用去离子水清洗4次,无水乙醇清洗3次,然后将洗净的产物放入80摄氏度的电热鼓风干燥箱中干燥24小时即得到了所要制备前驱体;
(5)称取70毫克的前驱体放入到石英舟内,另称取3克的升华硫放入另一石英舟内,然后将盛有前驱体的石英舟先推进管式炉加热区,再将盛有升华硫的石英舟推入同一个管式炉的加热区,并保证盛有升华硫的石英舟位置在盛有前驱体的石英舟的后面,使得高温升华后的硫蒸气在氮气的引流下经过前驱体,从而使前驱体进行硫化;
(6)将步骤(5)的管式炉中石英管内的空气排出,然后通入氮气,设置程序按照40摄氏度/分钟的升温速率升温至400摄氏度,保温时间设为2小时进行焙烧硫化,当温度降到室温时取出之前盛有前驱体的石英舟,其中的产物浸泡在去离子水中,并用超声机超声30分钟,然后再用去离子水清洗离心4遍,无水乙醇清洗离心3遍,将所得产物放入80摄氏度的烘箱中干燥12小时,该产物即是本发明所要制备的电催化剂材料。经XRD证明制备产物为MoS2/CoS2复合物,PDF卡片编号:37-1492和41-1471。
(7)将步骤(6)制备的产物制备成测试电极以测试电催化性能,制备方法如下:先将步骤(6)制备的产物在玛瑙研钵中进行研磨30分钟,然后取研磨后的产物7.5毫克于1.5毫升塑料离心管内,接着向该离心管内加入375微升的去离子水,然后将该盛有产物和溶剂的离心管用超声机超声5分钟后,再向该离心管中加入125微升无水乙醇和30微升的质量分数为5%nafion(全氟化树脂溶液)溶液,继续超声1小时,接着取超声完毕后的溶液13微升滴到3平方毫米采用300目三氧化二铝研磨抛光后的玻碳电极(Glass electrode简称“GC”)上,将该玻碳电极在室温下空气中自然干燥24小时后采用三电极法(铂片电极作为对电极;饱和银/氯化银电极作为参比电极;已制备好的玻碳电极作为工作电极)在法国Biologic电化学工作站上测试电催化裂解水的过电位以及交换电流密度等性能。
实施例2
(1)取0.291克六水合硝酸钴固体颗粒和0.121克脲素于反应容器中加入30毫升去离子水搅拌30分钟使反应物溶解完全;
(2)将步骤(1)中的溶液转移到带不锈钢衬底的100毫升的反应釜中,再将该反应釜放入已提前升温到140摄氏度的电热鼓风的烘箱中保持12小时,然后等反应釜温度降到室温时将反应釜中的上清液倒掉,底物采用去离子水清洗4次,无水乙醇清洗3次,然后将洗净的产物放入80摄氏度的电热鼓风干燥箱中干燥24小时即得到了所要制备二硫化钴的前驱体;
(3)称取70毫克的步骤(2)制备的前驱体放入到石英舟内,另称取3克的升华硫放入另一石英舟内,然后将盛有前驱体的石英舟先推进管式炉加热区,再将盛有升华硫的石英舟推入同一个管式炉的加热区,并保证盛有升华硫的石英舟位置在盛有前驱体的石英舟的后面,使得高温升华后的硫蒸气在氮气的引流下经过前驱体,从而使前驱体进行硫化;
(4)将步骤(3)的管式炉中石英管内的空气排出,然后通入氮气,设置程序按照40摄氏度/分钟的升温速率升温至400摄氏度,保温时间设为2小时进行焙烧硫化,当温度降到室温时取出之前盛有前驱体的石英舟,其中的产物浸泡在去离子水中,并用超声机超声30分钟,然后再进行用去离子水清洗离心4遍,无水乙醇清洗离心3遍,将所得产物放入80摄氏度的烘箱中干燥12小时,即是所要制备的CoS2电催化剂材料。经XRD证明制备产物为CoS2,PDF卡片编号:41-1471。
(5)将步骤(4)制备的产物制备成测试电极以测试电催化性能,制备方法如下:先将步骤(4)制备的产物在玛瑙研钵中进行研磨30分钟,然后取研磨后的产物7.5毫克于1.5毫升塑料离心管内,接着向该离心管内加入375微升的去离子水,然后将该盛有产物和溶剂的离心管用超声机超声5分钟后,再向该离心管中加入125微升无水乙醇和30微升的质量分数为5%nafion溶液,继续超声1小时,接着取超声完毕后的溶液13微升滴到3平方毫米采用300目三氧化二铝研磨抛光后的玻碳电极(Glass electrode简称“GC”)上,将该玻碳电极在室温下空气中自然干燥24小时后采用三电极法(铂片电极作为对电极;饱和银/氯化银电极作为参比电极;已制备好的玻碳电极作为工作电极)在法国Biologic电化学工作站上测试电催化裂解水的过电位以及交换电流密度等性能。
实施例3
(1)称取取0.242克二水合钼酸钠固体颗粒放入到石英舟内,另称取3克的升华硫放入另一石英舟内,然后将盛有前驱体的石英舟先推进管式炉加热区,再将盛有升华硫的石英舟推入同一个管式炉的加热区,并保证盛有升华硫的石英舟位置在盛有前驱体的石英舟的后面,使得高温升华后的硫蒸气在氮气的引流下经过前驱体,从而使前驱体进行硫化;
(2)将步骤(1)的管式炉中石英管内的空气排出,然后通入氮气,设置程序按照40摄氏度/分的升温速率加热温度设为400摄氏度,保温时间设为2小时进行焙烧硫化,当温度降到室温时取出之前盛有前驱体的石英舟,其中的产物浸泡在质量分数37%的浓盐酸中,并用超声机超声30分钟,然后再进行用去离子水清洗离心4遍,无水乙醇清洗离心3遍,将所得产物放入80摄氏度的烘箱中干燥12小时,即是所要制备的MoS2电催化剂材料。经XRD证明制备产物为MoS2,PDF卡片编号:37-1492。
(3)将步骤(2)制备的产物制备成测试电极以测试电催化性能,制备方法如下:先将步骤(2)制备的产物在玛瑙研钵中进行研磨30分钟,然后取研磨后的产物7.5毫克于1.5毫升塑料离心管内,接着向该离心管内加入375微升的去离子水,然后将该盛有产物和溶剂的离心管用超声机超声5分钟后,再向该离心管中加入125微升无水乙醇和30微升的质量分数为5%nafion溶液,继续超声1小时,接着取超声完毕后的溶液13微升滴到3平方毫米的采用300目三氧化二铝研磨抛光后的玻碳电极(Glass electrode简称“GC”)上,将该玻碳电极在室温下空气中自然干燥24小时后采用三电极法(铂片电极作为对电极;饱和银/氯化银电极作为参比电极;已制备好的玻碳电极作为工作电极)在法国Biologic电化学工作站上测试电催化裂解水的过电位以及交换电流密度等性能。
实施例4
(1)采用三电极法(铂片电极作为对电极;饱和银/氯化银电极作为参比电极;采用300目三氧化二铝研磨抛光的玻碳电极作为工作电极)在法国Biologic电化学工作站上测试玻碳电极的电催化裂解水的过电位以及交换电流密度等性能。
综上制备实例,我们得出如下进一步结论:
(1)采用该硫化法制备的三种电催化剂中在相同的测试条件下MoS2/CoS2复合物过电位与0点电位最近,并且交换电流密度的绝对值最大。
(2)升温速率和煅烧温度的对我们制备出所要的产物影响很大。
(3)产物的纯度对电催化性能测试的结果影响很大。
- 还没有人留言评论。精彩留言会获得点赞!