一种手性物质掺杂的聚偏氟乙烯共混超滤膜及其制备和应用的制作方法
本发明涉及对映体分离和膜分离技术领域,具体涉及一种手性物质掺杂的聚偏氟乙烯共混超滤膜的制备及其在色氨酸对映体分离中的应用。
背景技术:
随着药物合成技术的快速发展,具有光学活性的手性药物在医药、卫生领域中的应用日益增加,目前市场上使用的药物有近一半是手性药物,但手性药物大多是以外消旋体方式给药的,其对映体往往不具有药性,甚至还有较大的毒性。如氨基酸在实际的应用过程中,只有l-型氨基酸才能被利用,而摄入过量的d-型氨基酸则会引起中毒。因此手性药物的高效分离是目前药物研究中热点,也是药物生产中亟待解决的难题之一。
色谱法是目前对映体分离最主要方法,但该方法需要柱前衍生化,不仅费时,还可能使物质消旋化失去药性。其它的对映体分离方法还有结晶法、化学拆分法、酶拆分法等,但这些方法的分离效率都比较低,要消耗一种对映体;另外,化学拆分法还存在化学试剂难以回收,酶拆分法存在酶制剂不易获得等问题。膜分离技术在手性药物拆分中的应用已引起人们的极大关注。一些学者认为新型的膜分离技术是最有可能实现工业化的对映体药物拆分方法。
近年来,关于手性拆分膜及其对映体分离方面的研究日益增多,手性拆分膜分液膜和固膜两类。液膜的优点是通量大、选择性好,但主要缺点是稳定性差、使用寿命短。相对于液膜而言,固膜在稳定性方面已经达到了工业化生产的要求,但使用的膜基材往往不具有手性选择性、或手性选择性不高,需通过加入手性选择剂,也就是通过杂化来改善手性拆分膜的选择性。在外加的手性选择剂与膜基材的结合力较弱时,改性后的膜在使用过程中手性选择剂易流失,影响其稳定性及使用寿命;而当结合力较强时,会导致手性位点的失活。因此要提高手性固膜的分离性能和稳定性,膜中手性位点的数目其关键因素。
技术实现要素:
本发明提供了一种手性物质掺杂的聚偏氟乙烯共混超滤膜的溶液共混制备法,该法合成工艺简单,加入β-环糊精在乙酸纤维素中构成手性结构并引入大量手性位点,而后再将该手性复合材料与稳定性高的聚偏氟乙烯共混得到共混超滤膜。该共混超滤膜中的手性结构及其含量均可控。本发明制备得到的具有手性结构的纳滤复合膜可以对色氨酸对映体进行有效的拆分,通过改变反应条件参数或β-环糊精的加入量即可有效调控超滤复合膜对色氨酸对映体的分离效率。
一种手性物质掺杂的聚偏氟乙烯共混超滤膜的制备方法,包括如下步骤:
(1)将β-环糊精和乙酸纤维素溶解于n,n-二甲基二酰胺和丙酮的混合溶液中,水浴中搅拌均匀,超声后静置脱气泡后得到溶液a;将聚偏氟乙烯粉末和聚乙二醇溶解于n,n-二甲基乙酰胺中,水浴中搅拌均匀,而后超声后得到溶液b;
(2)将溶液a和溶液b在超声作用下混合,并升温至55~65℃均匀共混且充分反应,得到铸膜液;
(3)将所得铸膜液均匀涂布于位于玻璃平板上的基膜表面,成复合膜;经脱膜、干燥后得手性物质掺杂的聚偏氟乙烯共混超滤膜。
本发明以化学稳定和热稳定性能都非常高的聚偏氟乙烯作为膜材料组分,选择富含手性位点的乙酸纤维素和具有手性空间结构、且有较多手性位点的β-环糊精两种材料作为手性物质掺杂材料,通过溶液共混法制备手性物质掺杂的聚偏氟乙烯共混超滤膜,并将其用于色氨酸对映体的超滤分离过程。
优选地,步骤(1)中溶液a中β-环糊精的浓度为0.01~0.1g/ml、乙酸纤维素的浓度为0.05~0.5g/ml、n,n-二甲基二酰胺和丙酮的体积比为5.0:15.0;溶液b中聚偏氟乙烯的浓度为0.25~0.5g/ml、聚乙二醇浓度为0.08~0.1g/ml。
进一步优选地,溶液a中β-环糊精的浓度为0.05~0.1g/ml、乙酸纤维素的浓度为0.08~0.1g/ml。更进一步优选地,溶液a中β-环糊精的浓度为0.05~0.075g/ml、乙酸纤维素的浓度为0.1g/ml。该优选配比下,膜的通量和选择性都达到更好的状态。
进一步优选地,n,n-二甲基二酰胺和丙酮的体积比为5.0:15.0。
进一步优选地,溶液b中聚偏氟乙烯的浓度为0.3g/ml、聚乙二醇浓度为0.1g/ml。
乙酸纤维素为商品乙酸纤维素,通过市售获得,乙酰度20~50%,进一步优选为30~50%,最优选为40%;乙酸纤维素的羟基含量1~5%,进一步优选为1~3%,最优选为2%;乙酸纤维素分子量10000~30000以上,进一步优选为10000~20000,最优选为15000;聚乙二醇的分子量为400;聚偏氟乙烯为商品聚偏氟乙烯。上述步骤中所涉超声步骤超声时间均为半小时左右。
优选地,制备溶液a时的水浴温度为20~40℃、搅拌时间为5~10小时。进一步优选地,30℃搅拌6小时。
优选地,步骤(2)中溶液a与溶液b等体积混合;混合后的反应时间为1~3小时。进一步优选为1.5小时。
溶液a与溶液b等体积混合后升温至55~65℃进行反应,进一步优选升温至60℃进行反应。反应温度是来用来调控环糊精和醋酸纤维素与聚偏氟乙烯之间的共混效果,温度升高有利于形成均匀的共混铸膜液,有利于后续的共混膜的形成。
优选地,步骤(3)中,所述基膜为无纺布,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒至无纺布表面,将玻璃平板置于匀胶机上旋转成复合膜。
进一步地,匀胶机的转速为500~1000转/分钟。进一步优选为最优选800转/分钟。
进一步地,脱膜过程为:20~30℃(优选25℃)将带有复合膜的玻璃平板在蒸馏水中浸泡20~25小时后取出无纺布为基膜的β-环糊精/乙酸纤维素掺杂的聚四氟乙烯共混超滤膜材料;干燥条件为35~40℃下真空干燥,干燥时间为1~5小时。进一步优选,40℃下真空干燥4小时。
本发明中,可以简便地通过改变β-环糊精和乙酸纤维素比例和制备铸膜液的反应温度参数来调控复合共混超滤膜的手性结构和分离性能。
本发明还提供一种如所述制备方法制备得到的手性物质掺杂的聚偏氟乙烯共混超滤膜。
本发明还提供一种利用所述聚偏氟乙烯共混超滤膜分离色氨酸对映体的方法,包括如下步骤:
使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离,先将聚偏氟乙烯共混超滤膜在n2氛围下、操作压力为0.14~0.16mpa下以去离子水作为进料液预压25~35min,待其流速稳定后,将进料液换成d-色氨酸和l-色氨酸的混合水溶液,混合水溶液中含有乙酸,将压力调整为0.0.9~1mpa、实验温度24~26℃,进行分离。
进一步地,使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离,先将聚偏氟乙烯共混超滤膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离。
优选地,聚偏氟乙烯共混超滤膜面积为10~30cm2,膜厚度为10~50μm。进一步优选地,聚偏氟乙烯共混超滤膜面积为20cm2,膜厚度为25μm。此处的膜厚度是指去掉无纺布后的膜厚度,膜厚度通过控制匀胶机的转速进行调节。
优选地,所述分离实验原料液的体积为1~5l,优选为2l;原料液体中d-色氨酸和l-色氨酸浓度保持相同为0.1~2g/l,优选为1g/l;原料液中乙酸浓度为5~20g/l,优选为10g/l。
与现有技术相比,本发明具有如下有益效果:
本发明提供的手性物质掺杂的聚偏氟乙烯共混超滤膜材料以及及其制备方法简便有效,易于操作且成本较低。以聚偏氟乙烯为主要组分得到的共混超滤膜稳定性能优异。利用本发明的制备方法可以简便地通过改变β-环糊精和乙酸纤维素比例和反应温度以及刮膜条件等参数来调控复合共混超滤膜对手性物质的分离性能。
附图说明
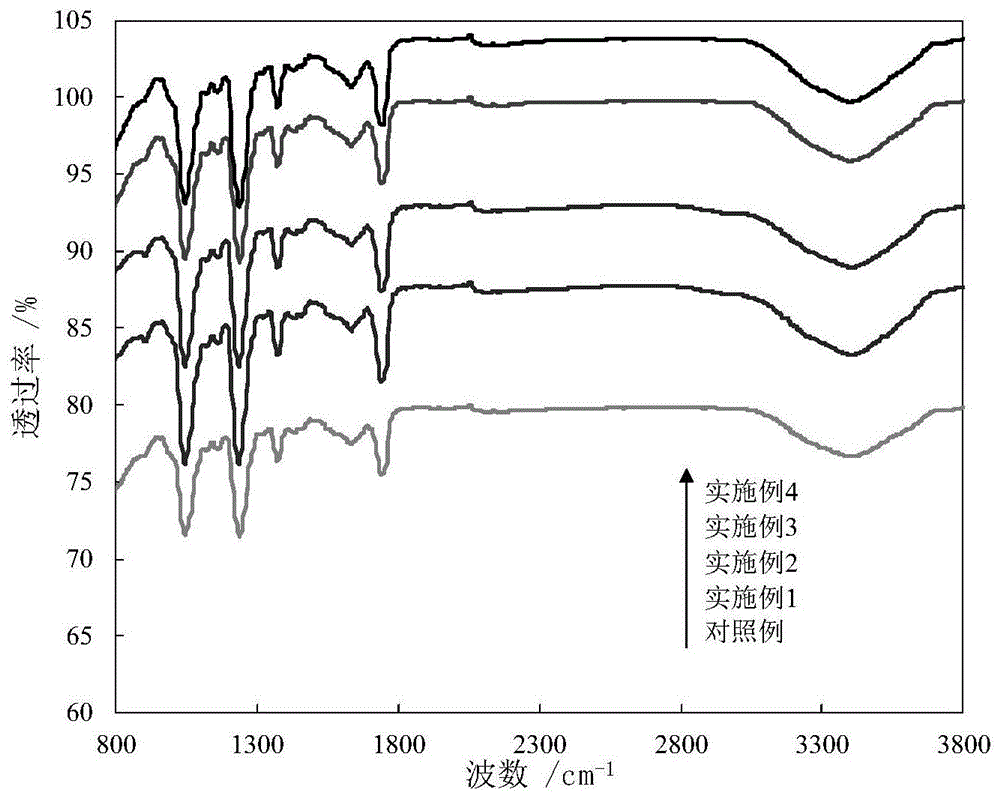
图1为本发明对照例和实施例所制备的纯聚偏氟乙烯膜和手性物质掺杂的聚四氟乙烯共混超滤膜的红外(ftir)图谱。
图2为本发明对照例和实施例所制备的纯聚偏氟乙烯膜和手性物质掺杂的聚四氟乙烯共混超滤膜的表面扫描电镜(sem)照片。
图3为本发明对照例和实施例所制备纯聚偏氟乙烯膜和性物质掺杂的聚四氟乙烯共混超滤膜的断面扫描电镜(sem)照片。
图4为本发明对照例和实施例所制备纯聚偏氟乙烯膜和性物质掺杂的聚四氟乙烯共混超滤膜的表面zeta电位图。
图5为本发明对照例和实施例所制备纯聚偏氟乙烯膜和性物质掺杂的聚四氟乙烯共混超滤膜的表面静态接触角测试图。
图6为本发明对照例和实施例所制备纯聚偏氟乙烯膜和性物质掺杂的聚四氟乙烯共混超滤膜的水通量图。
图7为本发明对照例和实施例所制备纯聚偏氟乙烯膜和性物质掺杂的聚四氟乙烯共混超滤膜的选择性图。
具体实施方式
下面介绍的为本发明较为优选的实施例,并不用于对本发明的限定。为了更好的对比具有手性结构和非手性结构膜结构和性能之间的差别,申请人还制备了不添加β-环糊精和乙酸纤维素的纯聚偏氟乙烯膜,作为空白膜进行对照。
对照例
(1)纯聚偏氟乙烯膜共混超滤膜的制备
将6.0g聚偏氟乙烯粉末溶解于20ml的n,n-二甲基二酰胺和2.0g的聚乙二醇(分子量400)的混合溶液中,30℃的水浴中搅拌5小时,而后超声作用半小时,静置脱气泡后得到铸膜液。以无纺布作为基膜,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒置无纺布表面,将玻璃平板置于匀胶机上控制800转/分钟的速率旋转成复合膜。在25℃下,将带有复合膜的玻璃平板在蒸馏水中浸泡24小时后取出纯聚偏氟乙烯膜,在40℃下真空干燥2小时得到纯聚偏氟乙烯膜。
(2)dl-色氨酸溶液的分离过程
在超滤装置中进行纯聚偏氟乙烯膜的色氨酸对映体分离实验,在实验室自制的超滤装置中,使用的纯聚偏氟乙烯膜的膜面积为20cm2、膜厚度为25μm。使用超滤装置进行乙酸纤维素共混超滤膜的色氨酸对映体分离实验,首先将膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离实验。其中原料液中d-色氨酸和l-色氨酸浓度保持相同为1g/l,原料液中乙酸为10g/l。利用高效液相色谱分析分离前后料液中d-色氨酸和l-色氨酸的含量,并以下列公式计算得到两者的选择性:
式中,ad和al分别代表透过液的高效液相色谱谱图中d-色氨酸和l-色氨酸的峰面积。
实施例1
(1)手性物质掺杂的聚偏氟乙烯共混超滤膜的制备
将0.2gβ-环糊精和2.0g乙酸纤维素溶解于20ml的n,n-二甲基二酰胺和丙酮的混合溶液中,30℃的水浴中搅拌5小时,而后超声作用半小时,静置脱气泡后得到溶液a。而后,将6.0g聚偏氟乙烯粉末和3.0g聚乙二醇(分子量为400)溶解于20mln,n-二甲基乙酰胺(dmac)烧杯中,水浴中搅拌均匀,而后超声作用半小时得到溶液b。将溶液a和溶液b在超声作用下混合,并升温至60℃均匀共混且充分反应,得到铸膜液。以无纺布作为基膜,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒置无纺布表面,将玻璃平板置于匀胶机上控制800转/分钟的速率旋转成复合膜。在25℃下,将带有复合膜的玻璃平板在蒸馏水中浸泡24小时后取出无纺布为基膜的聚偏氟乙烯共混超滤膜材料,在40℃下真空干燥2小时得到手性物质掺杂的聚偏氟乙烯共混超滤膜。
实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的红外图谱如图1所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面扫描电镜(sem)照片如图2所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的断面扫描电镜(sem)照片如图3所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面zeta电位如图4所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面静态接触角测试图如图5所示。
对比图1中实施例1的复合膜和对照例的纯聚偏氟乙烯膜可以发现,手性物质掺杂的聚四氟乙烯共混超滤膜的红外光谱图和纯聚偏氟乙烯膜的相似,两者的红外光谱图都在3320cm-1左右处出现-oh伸缩振动峰。图2膜的表面sem照片表明对比纯聚偏氟乙烯膜膜,β-环糊精和乙酸纤维素引入后复合膜表面的孔隙结构明显增多。两种膜的断面电镜照片显示,β-环糊精和乙酸纤维素的添加使手性膜的断面孔结构从海绵状孔向指状孔过渡,其孔隙半径明显增加。如图4和图5所示,β-环糊精和乙酸纤维素加入后使得复合膜的表面极性增加,其表面zeta电位值变大,表面亲水角也显著变小。
(2)聚偏氟乙烯共混超滤膜分离dl-色氨酸溶液的分离过程
在超滤装置中进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,在实验室自制的超滤装置中,使用的聚偏氟乙烯共混超滤膜的膜面积为20cm2、膜厚度为25μm。使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,首先将膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离实验。其中原料液中d-色氨酸和l-色氨酸浓度保持相同为1g/l,原料液中乙酸为10g/l。利用高效液相色谱分析分离前后料液中d-色氨酸和l-色氨酸的含量,并以下列公式计算得到两者的选择性:
式中,ad和al分别代表透过液的高效液相色谱谱图中d-色氨酸和l-色氨酸的峰面积。
实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的水通量如图6所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜分离色氨基酸的选择性如图7所示。
由图6和图7可以看出,β-环糊精和乙酸纤维素加入后共混超滤膜的通量和对色氨基酸的选择性都明显增加,显示了较好的分离效果。
实施例2
(1)聚偏氟乙烯共混超滤膜的制备
将0.5gβ-环糊精和2.0g乙酸纤维素溶解于20ml的n,n-二甲基二酰胺和丙酮的混合溶液中,30℃的水浴中搅拌5小时,而后超声作用半小时,静置脱气泡后得到溶液a。而后,将6.0g聚偏氟乙烯粉末和3.0g聚乙二醇(分子量为400)溶解于20mln,n-二甲基乙酰胺(dmac)烧杯中,水浴中搅拌均匀,而后超声作用半小时得到溶液b。将溶液a和溶液b在超声作用下混合,并升温至60℃均匀共混且充分反应,得到铸膜液。以无纺布作为基膜,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒置无纺布表面,将玻璃平板置于匀胶机上控制800转/分钟的速率旋转成复合膜。在25℃下,将带有复合膜的玻璃平板在蒸馏水中浸泡24小时后取出无纺布为基膜的聚偏氟乙烯共混超滤膜材料,在40℃下真空干燥2小时得到手性物质掺杂的聚偏氟乙烯共混超滤膜。
实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的红外图谱如图1所示;实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面扫描电镜(sem)照片如图2所示;实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的断面扫描电镜(sem)照片如图3所示;实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面zeta电位如图4所示;实施例1所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面静态接触角测试图如图5所示。
对比图1中实施例2的共混超滤膜和对照例的纯聚偏氟乙烯膜可以发现,手性物质掺杂的聚四氟乙烯共混超滤膜的红外光谱图和纯聚偏氟乙烯膜的相似,两者的红外光谱图都在3320cm-1左右处出现-oh伸缩振动峰。图2膜的表面sem照片表明对比纯聚偏氟乙烯膜,β-环糊精和乙酸纤维素引入后复合膜表面的孔隙结构明显增多。随着β-环糊精加入量增加,其孔隙数量明显增多。两种膜的断面电镜照片显示,β-环糊精和乙酸纤维素的添加使手性膜的断面孔结构从海绵状孔向指状孔过渡,其孔隙半径明显增加。且随着β-环糊精加入量增加,孔结构变化更为显著。如图4和图5所示,β-环糊精加入量增加后共混超滤膜的表面极性持续增加,其表面zeta电位值持续变小,表面亲水角也持续变小。
(2)聚偏氟乙烯共混超滤膜分离dl-色氨酸溶液的分离过程
在超滤装置中进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,在实验室自制的超滤装置中,使用的聚偏氟乙烯共混超滤膜的膜面积为20cm2、膜厚度为25μm。使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,首先将膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离实验。其中原料液中d-色氨酸和l-色氨酸浓度保持相同为1g/l,原料液中乙酸为10g/l。利用高效液相色谱分析分离前后料液中d-色氨酸和l-色氨酸的含量,并以下列公式计算得到两者的选择性:
式中,ad和al分别代表透过液的高效液相色谱谱图中d-色氨酸和l-色氨酸的峰面积。
实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的水通量如图6所示;实施例2所制备的手性物质掺杂的聚四氟乙烯共混超滤膜分离色氨基酸的选择性如图7所示。
由图6和图7可以看出,β-环糊精和乙酸纤维素加入后共混超滤膜的通量和对色氨基酸的选择性都明显增加,显示了较好的分离效果。随着β-环糊精加入量增加,手性物质掺杂的聚四氟乙烯共混超滤膜的通量和对色氨基酸的选择性都持续变大。
实施例3
(1)聚偏氟乙烯共混超滤膜的制备
将1.0gβ-环糊精和2.0g乙酸纤维素溶解于20ml的n,n-二甲基二酰胺和丙酮的混合溶液中,30℃的水浴中搅拌5小时,而后超声作用半小时,静置脱气泡后得到溶液a。而后,将6.0g聚偏氟乙烯粉末和3.0g聚乙二醇(分子量为400)溶解于20mln,n-二甲基乙酰胺(dmac)烧杯中,水浴中搅拌均匀,而后超声作用半小时得到溶液b。将溶液a和溶液b在超声作用下混合,并升温至60℃均匀共混且充分反应,得到铸膜液。以无纺布作为基膜,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒置无纺布表面,将玻璃平板置于匀胶机上控制800转/分钟的速率旋转成复合膜。在25℃下,将带有复合膜的玻璃平板在蒸馏水中浸泡24小时后取出无纺布为基膜的聚偏氟乙烯共混超滤膜材料,在40℃下真空干燥2小时得到手性物质掺杂的聚偏氟乙烯共混超滤膜。
实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的红外图谱如图1所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面扫描电镜(sem)照片如图2所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的断面扫描电镜(sem)照片如图3所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面zeta电位如图4所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的的表面静态接触角测试图如图5所示。
对比图1中实施例3的共混超滤膜和对照例的纯聚偏氟乙烯膜可以发现,手性物质掺杂的聚四氟乙烯共混超滤膜的红外光谱图和纯聚偏氟乙烯膜的相似,两者的红外光谱图都在3320cm-1左右处出现-oh伸缩振动峰。图2膜的表面sem照片表明对比纯聚偏氟乙烯膜,β-环糊精和乙酸纤维素引入后复合膜表面的孔隙结构明显增多。随着β-环糊精加入量增加,其孔隙数量明显增多。两种膜的断面电镜照片显示,β-环糊精和乙酸纤维素的添加使手性膜的断面孔结构从海绵状孔向指状孔过渡,其孔隙半径明显增加。且随着β-环糊精加入量增加,孔结构变化更为显著。如图4和图5所示,β-环糊精加入量增加后共混超滤膜的表面极性持续增加,其表面zeta电位值持续变小,表面亲水角也持续变小。
(2)聚偏氟乙烯共混超滤膜分离dl-色氨酸溶液的分离过程
在超滤装置中进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,在实验室自制的超滤装置中,使用的聚偏氟乙烯共混超滤膜的膜面积为20cm2、膜厚度为25μm。使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,首先将膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离实验。其中原料液中d-色氨酸和l-色氨酸浓度保持相同为1g/l,原料液中乙酸为10g/l。利用高效液相色谱分析分离前后料液中d-色氨酸和l-色氨酸的含量,并以下列公式计算得到两者的选择性:
式中,ad和al分别代表透过液的高效液相色谱谱图中d-色氨酸和l-色氨酸的峰面积。
实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的水通量如图6所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜分离色氨基酸的选择性如图7所示。
由图6和图7可以看出,β-环糊精和乙酸纤维素加入后共混超滤膜的通量和对色氨基酸的选择性都明显增加,显示了较好的分离效果。随着β-环糊精加入量增加,手性物质掺杂的聚四氟乙烯共混超滤膜的通量和对色氨基酸的选择性都持续变大。
实施例4
(1)聚偏氟乙烯共混超滤膜的制备
将1.5gβ-环糊精和2.0g乙酸纤维素溶解于20ml的n,n-二甲基二酰胺和丙酮的混合溶液中,30℃的水浴中搅拌5小时,而后超声作用半小时,静置脱气泡后得到溶液a。而后,将6.0g聚偏氟乙烯粉末和3.0g聚乙二醇(分子量为400)溶解于20mln,n-二甲基乙酰胺(dmac)烧杯中,水浴中搅拌均匀,而后超声作用半小时得到溶液b。将溶液a和溶液b在超声作用下混合,并升温至60℃均匀共混且充分反应,得到铸膜液。以无纺布作为基膜,将无纺布在蒸馏水中浸湿后,平铺于清洁的玻璃平板上,而后将铸膜液倒置无纺布表面,将玻璃平板置于匀胶机上控制800转/分钟的速率旋转成复合膜。在25℃下,将带有复合膜的玻璃平板在蒸馏水中浸泡24小时后取出无纺布为基膜的聚偏氟乙烯共混超滤膜材料,在40℃下真空干燥2小时得到手性物质掺杂的聚偏氟乙烯共混超滤膜。
实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的红外图谱如图1所示;实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面扫描电镜(sem)照片如图2所示;实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的断面扫描电镜(sem)照片如图4所示;实施例3所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的表面zeta电位如图4所示;实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的的表面静态接触角测试图如图5所示。
对比图1中实施例4的共混超滤膜和对照例的纯聚偏氟乙烯膜可以发现,手性物质掺杂的聚四氟乙烯共混超滤膜的红外光谱图和纯聚偏氟乙烯膜的相似,两者的红外光谱图都在3320cm-1左右处出现-oh伸缩振动峰。图2膜的表面sem照片表明对比纯聚偏氟乙烯膜,β-环糊精和乙酸纤维素引入后复合膜表面的孔隙结构明显增多。随着β-环糊精加入量增加,其孔隙数量明显增多。两种膜的断面电镜照片显示,β-环糊精和乙酸纤维素的添加使手性膜的断面孔结构从海绵状孔向指状孔过渡,其孔隙半径明显增加。且随着β-环糊精加入量增加,孔结构变化更为显著。如图4和图5所示,β-环糊精加入量增加后共混超滤膜的表面极性持续增加,其表面zeta电位值持续变小,表面亲水角也持续变小。
(2)聚偏氟乙烯共混超滤膜分离dl-色氨酸溶液的分离过程
在超滤装置中进行聚氟乙烯共混超滤膜的色氨酸对映体分离实验,在实验室自制的超滤装置中,使用的聚偏氟乙烯共混超滤膜的膜面积为20cm2、膜厚度为25μm。使用超滤装置进行聚偏氟乙烯共混超滤膜的色氨酸对映体分离实验,首先将膜在n2氛围下,操作压力为0.15mpa下以去离子水作为进料液预压半小时,待其流速稳定后,将进料液换成含有乙酸的d-色氨酸和l-色氨酸的水溶液,将压力调整为0.1mpa,实验温度25℃,进行分离实验。其中原料液中d-色氨酸和l-色氨酸浓度保持相同为1g/l,原料液中乙酸为10g/l。利用高效液相色谱分析分离前后料液中d-色氨酸和l-色氨酸的含量,并以下列公式计算得到两者的选择性:
式中,ad和al分别代表透过液的高效液相色谱谱图中d-色氨酸和l-色氨酸的峰面积。
实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜的水通量如图6所示;实施例4所制备的手性物质掺杂的聚四氟乙烯共混超滤膜分离色氨基酸的选择性如图7所示。
由图6和图7可以看出,β-环糊精和乙酸纤维素加入后共混超滤膜的通量和对色氨基酸的选择性都明显增加,显示了较好的分离效果。随着β-环糊精加入量增加,手性物质掺杂的聚四氟乙烯共混超滤膜的通量和对色氨基酸的选择性都持续变大。
以上所述仅为本发明专利的具体实施案例,但本发明专利的技术特征并不局限于此,任何相关领域的技术人员在本发明的领域内,所作的变化或修饰皆涵盖在本发明的专利范围之中。
- 还没有人留言评论。精彩留言会获得点赞!