一种催化柴油加氢转化与催化汽油加氢联合方法与流程
本发明涉及一种烃类加氢处理方法,具体的说是一种催化柴油加氢转化与催化汽油选择性加氢联合工艺方法。
背景技术:
进入新世纪以来,随着人们环保意识的日益增强、国家环保法规的日趋严格以及国民经济的快速发展,世界各国对清洁马达燃料的需求都在不断增加。催化裂化(FCC)技术是重油轻质化的主要工艺手段之一,在世界各国的炼油企业中都占有比较重要的地位。我国催化裂化装置年加工能力目前已经超过1亿吨,仅次于美国。在汽柴油品构成中,催化裂化汽油占80%左右,催化柴油占30%左右。近年来,随着国内所加工原油质量的日益重质化,催化裂化所加工的原料也日趋重质化和劣质化,加之许多企业为了达到改善汽油质量或增产丙烯的目的,对催化裂化装置进行了改造或提高了催化裂化装置的操作苛刻度,导致催化裂化的产品,特别是催化柴油的质量更加恶化。为提高石油资源的利用率,提高汽柴油燃料的整体质量水平,实现产品调合最优化和产品价值最大化的目标,满足国内对清洁燃料不断增长的需求,高芳烃柴油加氢转化生产高附加值石脑油组分和低硫清洁柴油燃料的加氢裂化新工艺技术具有很好的应用前景。国内外科研工作者也进行了大量的研究工作。国外已有采用加氢裂化工艺技术将催化裂化轻循环油转化为超低硫柴油和高辛烷值汽油调合组分的相关报道。如:1995年NPRA年会,DavidA.Pappal等人介绍了由Mobil、AkzoNobel/NipponKetjen和M.W.Kellogg公司开发的一种单段加氢裂化工艺技术;2005年NPRA年会,VasantP.Thakkar等人介绍了UOP公司开发的LCOUnicrackingTM技术。据报道,以上两种技术均可将低价值的催化循环油组分转化为高辛烷值汽油组分和优质柴油调合组分。但目前,现有技术也存在着一些问题,高芳烃柴油转化技术主要目的是将高芳烃柴油转化为高辛烷值汽油组分,同时,降低柴油的硫含量和改善柴油十六烷值,汽油组分和柴油组分的加氢存在着矛盾,当裂化段催化剂加氢能力过强时,裂化生成的汽油组分过度加氢,产品辛烷值较低,而裂化段催化剂加氢能力不足时,柴油产品质量较差,同时,会使得催化剂积碳速率加快,影响长周期运转。在此过程中,就涉及到了选择性加氢的问题,改善汽油组分与柴油组分的加氢选择性成为这类加氢裂化过程需要面对的问题。此外,催化汽油选择性加氢过程是高辛烷值汽油生产过程的另一个主要工艺技术,而该过程存在着反应压降上升迅速装置运转周期短及反应温升高致使催化汽油加氢过程辛烷值损失大的问题。中国专利201110321295.9公开了一种催化裂化和催化汽油加氢联合工艺方法,调整FCC装置分馏塔的操作条件,在分馏塔内对FCC汽油进行切割预分离,得到轻馏分和重馏分;轻馏分进行无碱脱臭,无碱脱臭后的轻馏分与热催化柴油一起进入加氢预分馏塔,分出轻汽油和中汽油;中汽油依次通过预加氢反应器和加氢脱硫反应器,进行缓和的选择性加氢,重馏分进行深度选择性加氢,所得两部分精制后馏分油与精制轻汽油混合,得到清洁汽油产品或调和组分。该发明过程利用催化柴油带走无碱脱臭过程形成的易结焦物质,有利于缓解FCC汽油加氢单元压降问题。但该方案影响了催化分馏装置的平稳运行,增加催化装置能耗,同时,催化汽油单独加氢过程,管线液相比例较少,不利于带走反应过程形成的生胶物质。中国专利02110319.4公开了一种降低催化裂化汽油硫含量的方法,该发明方法通过将部分催化重汽油组份掺入催化柴油中,进催化柴油加氢精制装置进行加氢精制处理,获取的加氢粗汽油作为催化重整原料进行催化重整处理,获得高辛烷值重整汽油。但是,由于重整装置对汽油进料杂质含量要求较为严格,催化汽油重汽油与柴油组分在苛刻操作条件下混合加氢,对催化裂化重汽油中原有的烯烃、芳烃等高辛烷值组分深度加氢饱和,再通过重整过程恢复芳烃,造成了催化裂化重汽油这部分高辛烷值组分的浪费。
技术实现要素:
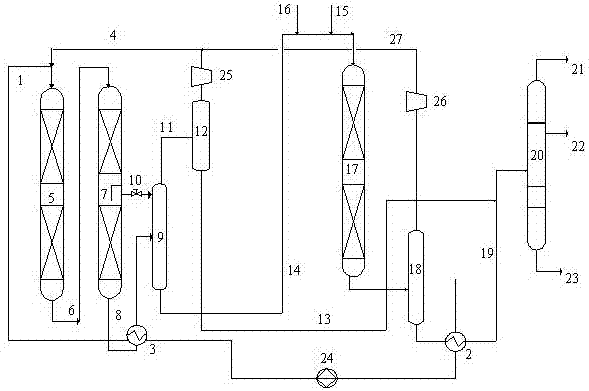
针对现有高辛烷值汽油生产过程中存在的问题,本发明提供了一种催化柴油加氢转化与催化汽油选择性加氢联合工艺方法,以改善高辛烷值汽油生产过程。本发明的一种催化柴油加氢转化与催化汽油选择性加氢联合方法,包括如下内容:(1)所述方法包括催化柴油加氢转化部分和催化汽油选择性加氢脱硫部分;(2)催化裂化柴油首先与催化汽油选择性加氢脱硫反应流出物进行换热,然后与氢气混合后与催化柴油加氢转化反应流出物进行换热,再经加热炉加热后进入加氢预处理反应器,在加氢精制条件下进行加氢精制反应;(3)加氢精制反应流出物不经分离直接进入加氢裂化反应器,与裂化反应器内级配装填的加氢裂化催化剂床层接触反应;所述催化剂的级配方式为:反应器内级配装填两种以上不同反应活性的加氢裂化催化剂,加氢裂化催化剂的加氢活性按照反应床层由上至下的顺序逐渐下降,加氢裂化催化剂的裂化活性由上至下逐渐降低;(4)步骤(3)的反应流出物进入热高分进行分离,得到气相与液相,气相进一步分离得到石脑油馏分和富氢气体;液相部分经减压后,与催化裂化汽油及新氢混合,不经加热直接进入催化汽油选择性加氢反应器,对催化汽油进行选择性加氢脱硫反应;(5)步骤(4)得到反应流出物进入分离器进行气液分离,得到气相经脱硫化氢处理后引入加氢转化的新氢压缩机入口,所得液相经分馏得到汽油和柴油产品。根据本发明的方法,其中步骤(4)中的催化汽油选择性加氢脱硫装置取消循环氢压缩机的设置。氢气管网引入的氢气(即新氢)进入催化汽油选择性加氢反应器反应后,经脱除硫化氢后直接进入催化柴油加氢转化部分的新氢压缩机入口,经升压后为催化柴油加氢转化装置反应补充氢气。根据本发明的方法,所述加氢裂化催化剂加氢活性的调整通过由上至下减少加氢裂化催化剂加氢活性金属组分含量的方式来实现。而加氢裂化催化剂的裂化活性可以通过调整催化剂中酸性分子筛的含量及分子筛的裂化活性来调整。按照床层由上至下,在裂化反应器内所使用的裂化催化剂中所含Y分子筛的含量逐渐下降;或者所含Y分子筛含量相同,而Y分子筛的裂化活性逐渐降低。所述的Y分子筛裂化活性主要指Y分子筛发生芳烃开环反应活性,作为裂化活组分,其裂化活性主要与Y分子筛红外酸量有关。可以通过调整Y分子筛中SiO2/Al2O3分子摩尔比来实现Y分子筛上红外酸量的调节,随着Y分子筛SiO2/Al2O3分子摩尔比的增大,红外酸量减少,Y分子筛活性裂化活性降低。在本发明方法中,选用的加氢精制催化剂和加氢裂化裂化剂可以使用市售产品,也可以根据本领域常规知识制备。本发明所用的加氢精制催化剂可采用常规加氢裂化预处理催化剂,一般以ⅥB族和/或第Ⅷ族金属为活性组分,以氧化铝或含硅氧化铝为载体。第ⅥB族金属一般为Mo和/或W,第Ⅷ族金属一般为Co和/或Ni。以催化剂的重量为基准,第ⅥB族金属含量以氧化物计为8wt%~28wt%,第Ⅷ族金属含量以氧化物计为2wt%~15wt%。所述的加氢裂化催化剂一般选择含有改性Y型分子筛的加氢裂化催化剂,以重量计催化剂含有WO315~30%,NiO2~15%,改性Y型分子筛30~90%,优选40~80%。所选用Y分子筛性质如下:比表面积700m2/g~900m2/g、总孔容0.35ml/g~0.48ml/g,相对结晶度90%~130%,晶胞参数2.437~2.460,红外酸量0.5~1.5mmol/g,载体通常为氧化铝或无定形硅铝。在本发明方法中,催化汽油选择性加氢部分选用的催化剂可以使用市售产品,也可以根据本领域常规知识制备。一般以氧化铝或含硅氧化铝为载体,Mo、Co为加氢活性组分。以催化剂的重量为基准,金属Mo含量以氧化物计为6wt%~20wt%,金属Co含量以氧化物计为1wt%~12wt%。本发明方法中,在加氢裂化反应器内由上至下,优选加氢裂化催化剂的孔径逐渐增大。裂化反应器内不同裂化催化剂可以按照等比例装填,也可以按照不同的比例装填。一般情况下,在级配装填的不同裂化催化剂床层,任意相邻的两种催化剂的比例为1:10~10:1。根据本发明的方法,还可以进一步包括以下内容:在步骤(3)中至少两个相邻的裂化催化剂床层之间设置气相引出管线,部分气相物流可以经由气相引出管线引出至催化柴油加氢转化部分高压分离器,而液相和剩余气相混合物继续进行加氢裂化反应。一般情况下,在反应条件下原料反应后气化率(气化率指在反应条件下,转化为气相的原料占原料总质量的比例,不含氢气)超过30质量%,优选超过50质量%的裂化催化剂床层间设置气相引出管线。所述的气相引出管线优选设置在催化剂床层间气液分配盘或冷氢箱的下方,气相引出管线安装有流量控制阀。正常操作时,气液分配盘或冷氢箱的下部会形成一个气相空间,气相引出管线在反应器内的开口设置在该气相空间中,液相物料基本不进入气相引出管线。本发明中优选在气相引出管线开口处设置折流档板,以进一步防止液相进入气相引出管线。在气相引出管线上面催化剂床层的氢油体积比一般为700:1~3000:1,优选800:1~1500:1;气相引出管线下面催化剂床层的氢油体积比一般为220:1~2000:1,优选300:1~1000:1。优选加氢裂化反应器的氢油比高于加氢精制反应器的氢油比200~800,最优选高出300~600。所述气相引出管线引出的气相物料量一般为循环氢气量(以体积计)的20%~70%,优选30%~60%。本发明方法中,所述的加氢精制反应的工艺条件包括:反应温度为320℃~440℃,优选340℃~420℃;反应压力为4.0MPa~15.0MPa,优选6.0MPa~12.0MPa;液时体积空速为0.2h-1~6.0h-1,优选0.5h-1~3.0h-1;氢油体积比为100~2000,优选500~1500。本发明中方法中,所述的催化汽油选择性加氢的工艺条件包括:反应温度为200℃~400℃,优选220℃~320℃;反应压力为0.5MPa~4.0MPa,优选1.0MPa~3.0MPa;液时体积空速为0.2h-1~10.0h-1,优选0.5h-1~6.0h-1;氢油体积比为10~500,优选30~300。本发明的方法中,所述催化裂化柴油的性质一般为:密度为0.88~0.99g/cm3,其干点一般为360~400℃,芳烃含量一般为50wt%~95wt%。催化裂化柴油的硫含量一般为0.2wt%~2wt%,氮含量为500μg/g~2000μg/g。催化裂化汽油的性质一般为:密度为0.70~0.80g/cm3,其干点一般为180~220℃,芳烃含量一般为5wt%~20wt%,烯烃含量一般为20wt%~50wt%。催化裂化汽油的硫含量一般为0.01wt%~0.2wt%,氮含量为10μg/g~200μg/g。与现有技术相比较,本发明的催化裂化柴油转化方法的优点是:1、在加氢裂化反应器内,由上至下随着裂化深度的加深,裂化反应器内反应物组分中裂解生成的石脑油含量由上至下逐渐增加,未裂化柴油部分由上至下逐渐下降。因此,采用本发明方法,裂化段催化剂床层加氢活性由上至下逐渐下降,在保证了催化裂化柴油加氢效果的同时,减少了石脑油的加氢,降低了氢耗,提高了石脑油产品辛烷值。此外,不同的加氢活性分布结合裂化活性从上至下逐渐降低,以及催化剂孔径的逐渐增大降低了反应生成的石脑油进一步裂解为轻烃的几率,提高了石脑油的收率。由于下部未转化催化柴油组分的裂化活性要高于反应生成的石脑油组分的裂化活性,且裂化反应器下部反应温度较高,因此,下部裂化催化剂较低的裂化活性在满足未转化的催化柴油进一步裂化的同时,降低了反应生成的石脑油的进一步裂化的几率,而下部裂化催化剂更大的孔径也降低了石脑油在催化剂上的扩散阻力,缩短了石脑油与裂化催化剂的接触时间,避免了反应生成的石脑油的进一步裂化,增加了石脑油的选择性。2、本发明方法在加氢裂化催化剂混合床层级配装填的基础上,在相邻的混合催化剂床层之间设置气相引出管线,同样可以将部分已经裂化的气相(主要包括气体轻和汽油组分)直接引出加氢裂化催化剂床层,也可以在一定程度上进一步减少了汽油的进一步加氢或裂化反应,一方面能够提高汽油的辛烷值,同时还可以提高加氢裂化的液体产品收率。3、通过提高裂化反应器的氢油比,更多地携带轻组分排出反应器,可进一步减少轻组分的二次或多次裂化反应,进而增加加氢裂化反应液收,提高汽油馏分的辛烷值。4、加氢裂化反应器下部催化剂床层中轻组分的二次或多次裂化反应减少,也有利于降低催化剂床层的积炭,延长装置的运转周期。5、催化汽油原料与催化柴油加氢转化部分未转化柴油混合后直接进入催化汽油选择性加氢脱硫反应器反应,与常规催化汽油选择性加氢工艺相比,催化汽油原料不经过换热器及加热炉加热,因此减少了换热器及加热炉内结焦的问题,另一方面未转化催化柴油馏分与催化汽油一起进入催化汽油选择性加氢脱硫装置反应,起到了吸收反应热的作用,降低了床层温升,避免了常规装置中底部过高温度对催化汽油过度加氢增加催化汽油辛烷值损失的问题。6、催化汽油选择性加氢脱硫部分取消循环氢压缩机的设置,直接利用管网氢气作为反应用氢,加氢转化部分与催化汽油选择性加氢装置共用一套分馏系统,与两套装置分别运转相比节省了投资。附图说明图1为本发明方法的一种原则工艺流程图。图2为本发明方法的另一种原则工艺流程图。具体实施方式下面结合附图和具体实施例对本发明的方法作更进一部的描述。如图1所示,催化裂化柴油首先进入催化汽油选择性加氢脱硫部分低压换热器2,与催化汽油选择性加氢脱硫装置流出物进行换热;然后,经升压泵24升压后进入催化柴油加氢转化部分的换热器3,与催化柴油加氢转化部分的反应流出物换热后经管线1与经管线4引入的氢气混合进入预处理反应器5进行加氢精制反应,精制反应器反应流出物经管线6直接进入裂化反应器7内与裂化反应器内级配的催化剂床层接触反应,反应流出物经管线8与经低压换热器2换热后的催化柴油原料在高压换热器3中换热后进入热高分9分离出气相和液相,气相部分经管线11进入分离器12分出石脑油馏分和氢气,分离所得的石脑油经管线13进入下游分馏塔20,分离得到的氢气经循环氢压缩机25和管线4循环回催化柴油预处理反应器;液相部分即未转化的催化柴油经减压后,经管线10与经管线15引入的低压新氢和经管线14引入的催化裂化汽油混合后直接进入催化汽油选择性加氢装置17对催化汽油进行选择性加氢脱硫反应,反应流出物进入分离器18进行气、液分离。分离得到的氢气经脱硫化氢处理后经新氢压缩机26加压后、经管线16循环回催化柴油加氢转化部分精制反应器入口作为催化柴油加氢转化部分的新氢使用,分离所得液相经换热器2和管线19,与加氢转化部分分离获得的石脑油馏分混合后进入分馏塔20,经分馏得到气体产品21、高辛烷值汽油22和低硫柴油组分23。如图2所示,本发明方法的另一种工艺流程为:催化裂化柴油首先进入催化汽油选择性加氢脱硫部分低压换热器2,与催化汽油选择性加氢脱硫装置流出物进行换热;然后,经升压泵24升压后进入催化柴油加氢转化部分的换热器3,与催化柴油加氢转化部分的反应流出物换热后经管线1与经管线4引入的氢气混合进入预处理反应器5进行加氢精制反应,精制反应器反应流出物经管线6直接进入裂化反应器7内与裂化反应器内级配的催化剂床层接触反应;在相邻的两个裂化催化剂床层之间,通过管线10引出部分气体至热高分9;反应流出物与经低压换热器2换热后的催化柴油原料在高压换热器3中换热后经管线8进入热高分9分离出气相和液相,气相部分经管线11进入分离器12分出石脑油馏分和氢气,分离所得的石脑油经管线13进入下游分馏塔20,分离得到的氢气经循环氢压缩机25、管线4循环回催化柴油预处理反应器;液相部分即未转化的催化柴油经减压后、经管线14与经管线15引入的低压新氢和经管线16引入的催化裂化汽油混合后直接进入催化汽油选择性加氢装置17对催化汽油进行选择性加氢脱硫反应,反应流出物进入分离器18进行气、液分离。分离得到的氢气经脱硫化氢处理后经新氢压缩机26加压后、经管线27循环回催化柴油加氢转化部分精制反应器入口作为催化柴油加氢转化部分的新氢使用,分离所得液相经换热器2和管线19与加氢转化部分分离获得的石脑油馏分混合后进入分馏塔20,分离出高辛烷值汽油22、低硫柴油组分23及气体产品21。本发明方法中,实施例中精制段采用常规的加氢预处理催化剂,通过调整精制段反应条件控制精制段流出油氮含量为30μg/g。裂化段分别选用了不同金属含量和不同分子筛含量的三种裂化催化剂的不同级配方式,裂化段三种不同裂化剂及催化汽油选择性加氢部分的催化剂的物化性质如下表1所示。下面通过实施例和比较例说明本发明的方案和效果。实施例1~3实施例1~3采用本发明的流程如图1。实施例1-3催化柴油加氢转化部分裂化反应器内按照催化剂床层由上至下的顺序分别级配装填表1中催化剂A、催化剂B、催化剂C三种裂化催化剂,在三个实施例中,催化剂A、催化剂B和催化剂C的比例分别为1:1:1;1:2:1和1:1:2。催化汽油选择性加氢部分催化剂选用Mo-Co型催化剂D。催化剂的组成及性质见表1,所用原料性质见表2,实施例1~3反应条件及反应结果见下表3和表4。实施例4采用图2所示流程。原料油、操作条件及所使用催化剂同实施例3。其中在第一和第二裂化催化剂床层之间设置气体引出管线,其他同实施例3。气相引出管线引出的气相物料量为循环氢气量的30%。实施例的工艺条件与结果分别列于表3和表4。表1实施例裂化催化剂的组成及主要性质。项目催化剂A催化剂B催化剂C催化剂D化学组成,质量%MoO3---12WO3242018CoO3NiO853载体氧化铝氧化铝氧化铝氧化铝裂化组分Y分子筛Y分子筛Y分子筛-裂化组分在载体中含量,wt%807060-裂化催化剂平均孔径,nm5.07.09.0-表2原料油性质。原料油催化柴油催化汽油密度,g/cm30.93240.7412馏程,℃192~35032~205S,wt%1.54320N,μg/g80040芳烃,wt%7210十六烷值18-辛烷值-92表3实施例1-4所采用操作条件。实施例1实施例2实施例3实施例4催化柴油转化部分反应温度,℃380390405405反应压力,MPa8.06.08.08.0体积空速,h-11.02.01.51.5氢油体积比1200120012001200裂化段进料氮含量,μg/g30303030单程转化率,wt%65658080化学氢耗,wt%3.323.373.643.30催化汽油选择性加氢部分平均反应温度,℃265290275275反应压力,MPa1.51.51.21.2体积空速,h-12.03.53.03.0氢油体积比300300200200表4实施例1-4评价结果。实施例1实施例2实施例3实施例4催化柴油加氢转化部分产品分布,wt%石脑油馏分馏分(<210℃)59587274柴油馏分(>210℃)35352020产品性质石脑油(<210℃)密度,g/cm-30.76060.76100.76020.7608S,μg/g<0.5<0.5<0.5<0.5N,μg/g<0.5<0.5<0.5<0.5辛烷值929391.892.2柴油(>210℃)密度,g/cm-30.88020.88300.87500.8748硫含量,μg/g71055十六烷值27303334催化汽油选择性加氢部分密度,g/cm-30.73890.73850.74000.7402馏程,℃30~20530~20530~20530~205硫含量,μg/g4081818辛烷值91.490.391.992.1比较例1-4比较例1-4催化柴油转化部分采用与实施例1-3相同的工艺条件,比较例1-4加氢预处理反应器内装填与实施例1-3中相同的预处理催化剂,并通过控制操作条件调整精制段流出油的氮含量为30μg/g,裂化段催化剂比较例1、比较例2中装填催化剂A,比较例3、比较例4中装填催化剂C。比较例1-4中使用的原料油与实施1-3相同。比较例1-2催化汽油选择性加氢部分分别控制与实施例1和实施例2相同的产品硫含量,调整反应条件。与比较例1-4操作条件及产品性质列于表5、表6。表5比较例1-4所采用操作条件。比较例1比较例2比较例3比较例4催化柴油加氢转化部分反应温度,℃372398390415反应压力,MPa8.08.08.08.0体积空速,h-11.01.51.01.5氢油体积比1200120012001200裂化段进料氮含量,μg/g30303030单程转化率,wt%65803580化学氢耗,wt%3.724.163.303.58催化汽油选择性加氢部分平均反应温度,℃210300反应压力,MPa1.51.5体积空速,h-12.03.5氢油体积比300300表6比较例1~4试验结果。比较例1表较例2比较例3比较例4产品分布,wt%石脑油馏分馏分(<210℃)57695465柴油馏分(>210℃)35203520产品性质石脑油(<210℃)密度,g/cm-30.76000.76020.76000.7602S,μg/g<0.5<0.5<0.5<0.5N,μg/g<0.5<0.5<0.5<0.5辛烷值88869291.5柴油(>210℃)密度,g/cm-30.88020.87500.88020.8750硫含量,μg/g7575十六烷值29342528催化汽油选择性加氢部分密度,g/cm-30.74380.7435馏程,℃29~20528~205硫含量,μg/g397.0辛烷值90.588.4催化柴油加氢转化部分通过实施例1、实施例3与比较例1和比较例2对比发现,在采用相同工艺条件加工表2中原料时,比较例1和比较例2使用高加氢活性和裂化活性的裂化剂A,在控制与实施例相同转化率的情况下,由于比较例1和比较例2催化剂较高的加氢活性使得比较例1和比较例2在柴油性质及石脑油收率与实施例1和实施例3相近的情况下,反应氢耗要明显高于实施例1和实施例3,其化学氢耗分别增加0.4和0.5个单位,而且生成的汽油组分辛烷值分别降低4和5.8个单位。比较例3和比较例4使用低加氢活性和低裂化活性的裂化剂C,结果在达到与实施例1和实施例3相同转化率的情况下,反应温度提高10℃,过低的裂化活性及过高的反应温度促进了二次裂解反应进行,使得重石脑油收率分别比实施1和实施例3降低5个和7个单位,而石脑油辛烷值及产品氢耗方面也没有明显的改善。总体来看催化柴油转化部分实施例相比比较例表现出了很好裂化及加氢选择性,使用效果明显好于比较例。催化汽油选择性加氢部分比较结果表明,比较例1和比较2与实施例1和实施例2相比,达到相同的脱硫深度时,由于温升较大致使其平均反应温度相同的情况下,出口温度更高,因此,加大了辛烷值损失,产品辛烷值损失明显高于实施例,产品硫含量控制与实施例相当时,辛烷值损失分别比实施例1和实施例2高出0.9个单位和1.9个单位。此外,相比于实施例3,实施例4通过在裂化床层间抽出裂化生成的石脑油,避免了石脑油在裂化反应器下部的进一步裂化和加氢饱和,因此,相比于实施例3,实施例4催化柴油加氢转化部分重石脑油收率及辛烷值明显增加。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1