一种基于等重二甲基化标记的多重蛋白质定量方法与流程
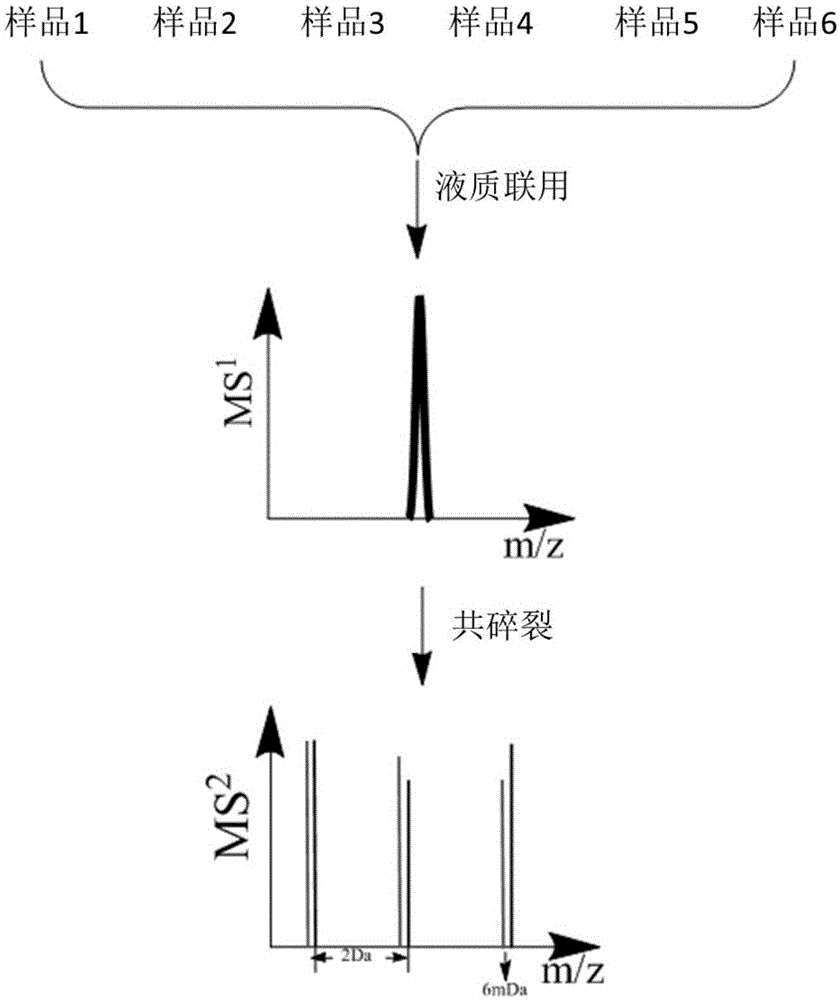
本发明涉及一种等重多重标记定量方法,可实现最多六重蛋白质样品的同时大规模定量。该方法具有标记效率高,标记选择性好,定量准确度、精密度、覆盖率高,动态范围宽,蛋白质组定量分析的通量高等优点。可以用于大样品量生物样品、生物体动态过程的蛋白质组定量分析。
背景技术:
蛋白质含量的变化可以反映生命过程、揭示疾病状态,探究该变化可以为寻找药物靶点和潜在疾病标志物提供基础数据支持。针对蛋白质组样品数量大、含量会随时间和空间发生变化等特点,在保证定量准确度、精密度、动态范围的前提下,亟需发展能够实现多重蛋白质组样品同时分析的定量方法。
目前多重蛋白质组定量方法常使用基于液相色谱质谱联用的稳定同位素标记策略,按照质谱中定量信息所在位置可以分为基于一级谱的定量方法和基于二级谱的定量方法。基于一级谱的定量方法通过比较多重标记样品在质谱一级谱中的峰强度或者谱峰面积实现。目前可以实现SILAC三重标记(Nat.Protoc.2011,6,147.)、SILAC结合二甲基化六重标记(Scientific Reports 2013,3,1827.)、二甲基化五重标记(Chem.Commun.2014,50,1708.)等。但这些方法不能避免一级谱噪音大、灵敏度低、数据解析困难、多重标记一级谱图复杂度成倍增加等不足。
相比上述方法,基于二级谱的定量方法由于多重标记一级谱质荷比相同,因此降低了一级谱图复杂度,有利于定量的深度覆盖;且二级谱信噪比明显提高,有利于提高定量准确度和动态范围。基于二级谱的定量方法中,基于报告离子的定量方法可以完成iTRAQ八重标记(Proteomics,2007,7,3651.)、TMT六重标记(Anal Chem,2008,80,2921.),但存在低估效应,影响定量准确度。近来基于碎片离子的定量方法越来越受到关注,目前已有IPTL三重标记(Anal.Chem.2013,85,2478.)、质量亏损四重标记(Anal.Chem.2013,85,10658.),该种方法定量准确度高、精密度好,动态范围更宽,但目前通量较小。
技术实现要素:
本发明发展了一种基于等重二甲基化标记的多重蛋白质定量方法,结合IPTL和基于质量亏损的标记方法,对Lys-C酶解的肽段进行不同pH条件下两端的二甲基化标记,实现等重六重标记,利用二级谱碎片离子进行定量。
为了实现上述目的,本发明采用的技术方案为:
1、蛋白质样品经过变性还原烷基化后,按照一定的酶/蛋白比例加入Lys-C酶,孵育过夜。
2、根据肽段的N末端氨基和C末端赖氨酸侧链氨基在酸性条件下反应速度不同的性质,将一份酶解后的蛋白样品首先在酸性条件下选择性二甲基化标记 肽段末端的氨基;然后在碱性条件下相应的二甲基化标记肽段赖氨酸上的氨基。酸性条件控制在pH值为2.0~5.0,标记时间5~120min;碱性条件控制pH值为7.5~12,标记时间5~120min。
3、重复上述步骤,进行两重、三重、四重、五重、六重样品标记。
4、上述多重标记方法中,六重标记试剂选择为:
肽段N末端:13CH2O+NaBH3CN——肽段C末端:CD2O+NaBD3CN;
肽段N末端:CD2O+NaBH3CN——肽段C末端:13CH2O+NaBD3CN;
肽段N末端:13CD2O+NaBH3CN——肽段C末端:CH2O+NaBD3CN;
肽段N末端:CH2O+NaBD3CN——肽段C末端:13CD2O+NaBH3CN;
肽段N末端:13CH2O+NaBD3CN——肽段C末端:CD2O+NaBH3CN;
肽段N末端:CD2O+NaBD3CN——肽段C末端:13CH2O+NaBH3CN。
5、将多重标记样品混合,进行后续的液质分析。液质联用中的质谱仪包括静电场轨道阱(Orbitrap)、飞行时间管(TOF)以及傅立叶变换离子回旋共振质量分析器(FT-ICR)。
6、采集到的肽段在质谱仪中得到的一级谱具有相同的质荷比,而二级谱每种碎片离子都存在质荷比差异,利用多重标记在每张二级谱上都同时出现,提取每种标记肽段在二级谱上六重标记都同时出现的a、b、y碎片离子强度并加和,作为该种标记肽段的强度。将每重强度相比作为相对定量结果,实现基于二级谱碎片离子强度的多重定量分析。
本发明具有如下优点:
1、标记效率高,标记选择性好:使用优化的标记条件,其肽段标记效率和标记选择性均在95%以上,且标记试剂价格低廉。
2、多重标记之间引入的同位素原子相同,不会因引入不同数目的氘原子而使多重肽段在反相色谱保留时间上存在差异。
3、定量通量大,可以实现六重样品的同时分析,大大缩短了样品分析的时间,提高了定量准确性。
4、标记方法中引入质量亏损的标记思路,有效地降低了二级谱图复杂程度,利于二级谱鉴定。
5、定量覆盖率、准确度、精密度高。
6、定量动态范围宽。
附图说明
图1等重二甲基化标记原理示意图;
图2六重标记样品混合理论质谱图示意图;
图3六重样品等量混合定量结果图,图为六重样品按1:1:1:1:1:1混合后定量比值取log2后所得。
具体实施方式
实施例1
1.蛋白质酶解
蛋白样品溶于8M尿素溶液中,加入二硫苏糖醇至终浓度为10mM,56℃变 性还原1小时,加入碘乙酰胺至终浓度为20mM,避光反应30min,再将尿素稀释至1M,按照1:50(酶/蛋白,w/w)加入Lys-C,37℃酶解过夜。用C18捕集柱除盐蒸干,用水复溶。
2.肽段二甲基化标记
对肽段分别进行六重二甲基化标记,如表3所示,具体标记方法如下:
第一重标记:将酶解后的肽段上样至C18捕集柱上,首先用酸性溶液体积浓度1.5%醋酸水溶液平衡捕集柱。在酸性条件下选择性二甲基化标记肽段末端的氨基,标记溶液为在1mL体积浓度1.5%醋酸的水溶液中加入50μL体积浓度4%13CH2O、50μL质量浓度0.6M NaBH3CN,流速50μL/min,标记20min。再依次使用酸性溶液体积浓度1.5%醋酸的水溶液、水冲洗捕集柱后用碱性溶液pH 8.0磷酸盐缓冲溶液平衡捕集柱。在碱性条件下二甲基化标记肽段赖氨酸上的氨基,标记溶液为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%CD2O、50μL质量浓度0.6M NaBD3CN,流速50μL/min,标记10min。用碱性溶液pH 8.0磷酸盐缓冲溶液、水依次冲洗捕集柱;最后用体积浓度80%ACN将样品从捕集柱上洗脱。
第二重标记:与第一重相同的标记方法,酸性条件下标记溶液替换为在1mL体积浓度1.5%醋酸的水溶液中加入50μL体积浓度4%CD2O、50μL质量浓度0.6M NaBH3CN,碱性条件下标记溶液替换为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%13CH2O、50μL质量浓度0.6M NaBD3CN。
第三重标记:与第一重相同的标记方法,酸性条件下标记溶液替换为在1mL体积浓度1.5%醋酸的水溶液中加入50μL体积浓度4%13CD2O、50μL质量浓度0.6M NaBH3CN,碱性条件下标记溶液替换为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%CH2O、50μL质量浓度0.6M NaBD3CN。
第四重标记:与第一重相似的标记方法,将酶解后的肽段上样至C18捕集柱上,首先用酸性溶液体积浓度1.5%醋酸的D2O溶液平衡捕集柱。在酸性条件下选择性二甲基化标记肽段末端的氨基,标记溶液为在1mL体积浓度1.5%醋酸的D2O溶液中加入50μL体积浓度4%CH2O、50μL质量浓度0.6M NaBD3CN,流速25μL/min,标记30min。再依次使用酸性溶液体积浓度1.5%醋酸的水溶液、水冲洗捕集柱后用碱性溶液pH 8.0磷酸盐缓冲溶液平衡捕集柱。在碱性条件下二甲基化标记肽段赖氨酸上的氨基,标记溶液为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%13CD2O、50μL质量浓度0.6M NaBH3CN,流速50μL/min,标记10min。用碱性溶液pH 8.0磷酸盐缓冲溶液、水依次冲洗捕集柱;最后用80%ACN将样品从捕集柱上洗脱。
第五重标记:与第四重相同的标记方法,酸性条件下标记溶液替换为1mL体积浓度1.5%醋酸的D2O溶液中加入50μL体积浓度4%13CH2O、50μL质量浓度0.6M NaBD3CN,碱性条件下标记溶液替换为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%CD2O、50μL质量浓度0.6M NaBH3CN。
第六重标记:与第四重相同的标记方法,酸性条件下标记溶液替换为1mL体积浓度1.5%醋酸的D2O溶液中加入50μL体积浓度4%CD2O、50μL质量浓 度0.6M NaBD3CN,碱性条件下标记溶液替换为在1mL pH 8.0磷酸盐缓冲溶液中加入50μL体积浓度4%13CH2O、50μL质量浓度0.6M NaBH3CN。
用冷冻蒸干仪干燥每重样品后用A相(含体积浓度2%ACN和体积浓度0.1%FA的水溶液)复溶至质量浓度0.5mg/mL。将样品按比例混合,待质谱分析。
3.LC-MS分析
液相色谱条件:流动相,缓冲液A相(含体积浓度2%ACN和体积浓度0.1%FA的水溶液),缓冲液B相(含体积浓度98%ACN和体积浓度0.1%FA的水溶液),样品分离梯度为0到10min,为0%B,然后进行125min从5%B到25%B的线性梯度分离,再进行10min从25%到35%B的线性梯度分离,流速为300nL/min。
质谱条件:使用Q Exactive,采用DDA模式,Full MS分辨率为70000(m/z=200),质量范围350-1800m/z;MS/MS选择最强的10个离子进行碎裂,动态排除20.0s,碎裂模式为HCD,归一化碰撞能量为28%,分离窗口2.0Da,MS2起始分子量为50.0Da。MS/MS分辨率为35000(m/z=200)。上述每个样品平行进样三次。
4.数据分析
质谱采集的原始文件使用MaxQuant(v1.2.2.5)软件包,并使用Andromeda作为搜库引擎进行数据库搜索。固定修饰:C为Carbamidomethyl,可变修饰:Acetyl(Protein N-term);Oxidation(M),N-term和K分别选择相应的二甲基化标记修饰。人类蛋白质数据库下载于ftp.uniprot.org(2013年7月)。一级谱质量容差6ppm,二级谱质量容差20ppm,蛋白质酶为Lys-C,允许两个漏切位点,肽段和蛋白质的FDR<0.1%。定量分析时,对鉴定到的肽段进行理论碎裂并与对应的mgf文件进行匹配,提取所有符合的碎片离子。然后进行离子对匹配,保留每重都出现的碎片离子,再将所有该标记的多种碎片离子强度加和作为该肽段的离子强度,多重标记肽段的离子强度再两两相比,作为该谱图鉴定到肽段的多重相对定量比值。相同的肽段取中位数作为该肽段的定量结果,相同的蛋白取所有鉴定到的肽段的中位数作为该蛋白的定量结果。三次重复实验,将所有结果合并到一起,相同的蛋白质取平均数,作为蛋白质的最终定量结果。所有的分析由SPSS(v20)和Excel(v2010)完成。
方法评价:
1、六重样品按1:1:1:1:1:1混合,定量覆盖率大于95%,实现了六重样品同时定量分析,定量准确度和精密度见表1、图3,可见该方法定量结果与理论值相符,精密度高。
2、定量动态范围宽:将样品按照1:2:5:10:20:50混合,结果如表2所示,本方法可以实现50倍的动态范围,定量准确度依然很好,不存在基于报告离子定量方法的低估效应。这得益于本方法是基于二级谱多种碎片离子进行定量的,可以有效降低共洗脱组分对定量的干扰。
实施例2
标记过程在离心管中进行,酸性标记后经过捕集柱进行溶剂交换,洗脱后进行碱性标记,其他过程同实施例1。
实施例3
酸性标记过程在离心管中进行,然后经过捕集柱进行溶剂交换,在捕集柱上进行碱性标记,其他过程同实施例1。
实施例4
在酸性条件下对肽段的N端氨基进行选择性二甲基化标记的标记溶液组成为浓度1%醋酸水溶液、4%甲醛水溶液及其同位素形式、0.6M氰基硼氢化钠溶液及其同位素形式,标记时间为10min,其他过程同实施例1。
实施例5
实施例1中选择前四种标记方法,即实现四重标记,其他过程同实施例1.
表1 六重等量混合定量准确度、精密度结果
表2 动态范围定量结果
表3 六重标记方法表
该方法的优势在于:定量准确度高,精密度好,动态范围宽,可以同时实 现六重蛋白质样品的同时定量分析,大大提高蛋白质定量分析的通量,节约分析时间。
- 还没有人留言评论。精彩留言会获得点赞!