一种分离测定唾液中吡嗪类和吡啶类物质的方法与流程
本发明属于分析化学技术领域,具体涉及一种专用于分离测定唾液中吡嗪类和吡啶类物质的方法。
背景技术:
吡嗪类物质是1、4位含两个杂氮原子的杂环类化合物,一般具有烤香、坚果香、爆米花香、咖啡香等香味特征。吡啶类物质是含有一个氮杂原子的六元杂环化合物,一般具有青香、木香、琥珀香等香味特征。吡嗪类和吡啶类物质是食品、烟草等中的重要香味成分。
饮食过程中食品中化学成分被口腔唾液选择性溶解吸收,那些被吸收的成分才是刺激人体产生感官感受的物质,决定了食品的风格和风味。目前对食品香味的评价通常以口、鼻、舌、喉等的感知来评价,但很难排除人为主观性以及由此所产生的不确定性。研究测定唾液中吸收溶解的风味成分,则能直接、客观的得到决定食品风味的化学成分数据。进行饮食者唾液中吡嗪类、吡啶类香吃味成分的分析,可为食品、烟草等的香吃味评定提供数据支持,为改善食品风味提供理论依据。
目前,关于香精香料、食品、白酒、烟气等样品中吡嗪类和吡啶类物质的测定已有大量的报道,然而唾液样品中吡嗪类和吡啶类物质的测定还未见报道。唾液样品基体复杂,主要成分是水(占99%),但含有多种有机物如黏蛋白、黏多糖、唾液淀粉酶、溶菌酶、免疫球蛋白、血型物质、尿素、尿酸和游离氨基酸等,以及无机物和一些气体分子,被分析成分在唾液中含量极低,还会受到唾液中多种有机成分和无机成分的干扰。因此,开发简单快速、高选择性、高灵敏度、定量准确的唾液中吡嗪类和吡啶类物质的提取和分离测定方法非常迫切。
技术实现要素:
本发明的目的在于针对目前唾液中吡嗪类和吡啶类物质分离测定的方法未见文献报道的现状,以及现有技术的不足,提供一种分离测定唾液中吡嗪类和吡啶类物质的方法,该方法可行,定性准确,精密度好,检出限低,准确可靠,具有推广应用价值。
为了实现上述目的,本发明采用的技术方案如下:
一种分离测定唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将脱脂棉置于口腔内,放置0.2-5 min后,取出吸收了唾液的脱脂棉,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品;
步骤(2),萃取:移取唾液样品于离心管中,加入萃取溶剂、盐和除水剂,经涡旋振荡至混合均匀后离心,取萃取液层,取萃取液层,得到萃取液,所述的萃取液中不含水;
所述的萃取溶剂为二氯甲烷、三氯甲烷、甲苯、乙酸乙酯和乙腈中的一种或几种的组合物(当为组合物时,具体比例没有限制),萃取溶剂加入量与唾液样品的体积比为0.2-2:1;所述的盐为氯化钠、柠檬酸钠和柠檬酸氢二钠中的一种或几种的组合物(当为组合物时,具体比例没有限制),盐的加入质量为唾液样品质量的10-40%;所述除水剂为无水硫酸镁、无水硫酸钠中的一种或两种的组合物(当为组合物时,具体比例没有限制),除水剂加入质量为唾液样品质量的0-70%。
步骤(3),测定:将步骤(2)收集到的萃取液过0.22 μm微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,外标法定量;
其中,气相色谱条件如下:
色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱或相当者;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;分流进样或不分流进样,分流比1-30;进样量:1-2 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃/min 升温到280℃,保持5 min。
飞行时间质谱条件如下:
电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为160-220℃,电离能量为40-60 eV,溶剂延迟:4.0-6.0 min,反应气为甲烷,流量为40%-60%,采集方式为总离子流色谱图(TIC),监测的特征离子参数见表1。
表1 吡嗪类和吡啶类监测的特征离子
进一步,优选的是步骤(1)中的脱脂棉的质量为0.1-2.0 g。
进一步,优选的是步骤(2)中唾液移取量为0.5-10 mL。
进一步,优选的是步骤(2)中涡旋振荡转速为100-20000 rpm,涡旋振荡时间为0.2-10 min,离心转速不低于10000 rpm,离心时间不少于5 min。
进一步,优选的是步骤(2)中,对取萃取液层后留下的溶液再加入萃取溶剂,重复与步骤(2)相同的萃取方式萃取,萃取后合并萃取液层,每次萃取溶剂的用量与唾液样品的体积比为0.2-2:1;重复萃取的次数为1-3次。
进一步,优选的是步骤(3)中萃取液需浓缩后再过微孔滤膜,浓缩的方法为减压蒸发(压力没有限制),减压蒸发的温度为20-50℃,浓缩至1-2 mL。
进一步,优选的是所述的吡嗪类物质为2-甲基吡嗪、2,6-二甲基吡嗪、2,3-二甲基吡嗪、三甲基吡嗪和四甲基吡嗪;所述的吡啶类物质为吡啶、3-乙基吡啶和5-乙基-2-甲基吡啶。
进一步,优选的是配制吡嗪类和吡啶类物质系列标准溶液,浓度为0.05 mg/L、0.1 mg/L、0.5 mg/L、1 mg/L、2 mg/L、5 mg/L、10 mg/L,对系列标准溶液进行气相色谱-飞行时间质谱联用仪分析,得到标准溶液的总离子流色谱图(如图1所示),根据各测定物质特征离子峰面积为纵坐标,以各测定物质浓度为横坐标,作各测定物质的标准曲线;根据标准曲线计算样品中吡嗪类和吡啶类物质含量,以两次平行测定的平均值为最终测定结果。
本发明与现有技术相比,其有益效果为:
本发明方法可以有效的用于测定唾液中吡嗪类和吡啶类物质,方法可行,前处理简单,操作步骤简化,定性鉴定准确,检测限低,精密度好,准确可靠,具有推广应用价值。
以上说明为本发明技术方案的概述,为了更清楚的了解本发明的技术手段,而可依照说明书的内容予以实施,并且为了让本发明的上述和其它目的、特征和优点能够更明显易懂,现特举较佳实施例,并配合附图,详细说明如下。
附图说明
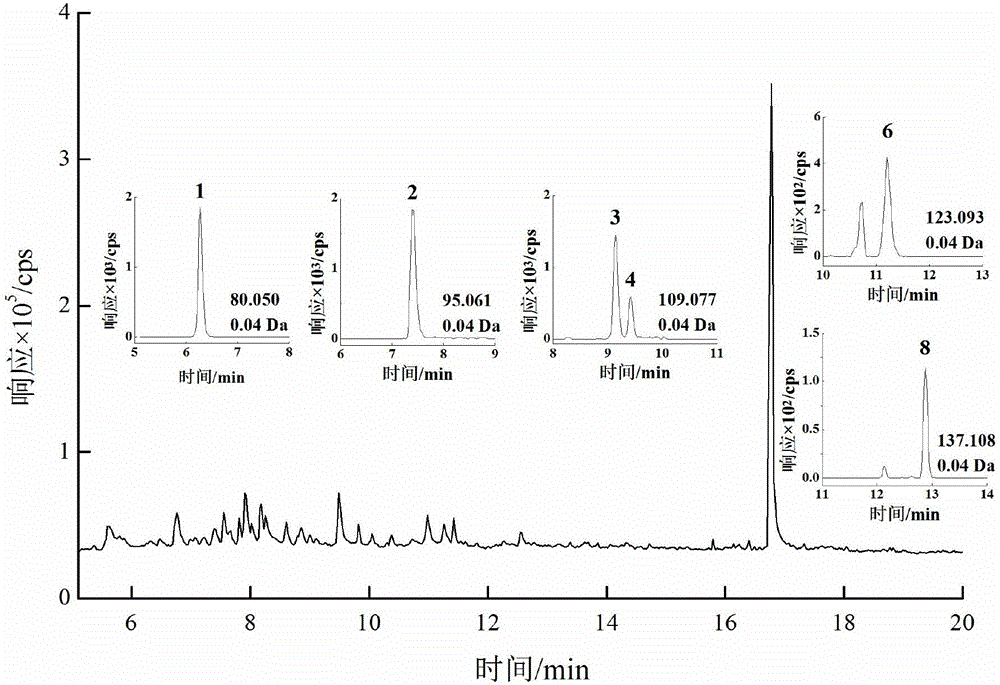
图1是本发明吡嗪类和吡啶类物质混合标准溶液(1.0 mg/L)的总离子流色谱图;
图2为烟草吸食者唾液样品的总离子流图和提取离子流图;
图3为白酒饮用者唾液样品的总离子流图和提取离子流图。
图中各峰的含义如下:1,6.26 min,吡啶;2,7.29 min,2-甲基吡嗪;3,9.18 min,2,5-二甲基吡嗪;4,9.41 min,2,3-二甲基吡嗪;5,10.76 min,3-乙基吡啶;6,11.15 min,三甲基吡嗪;7,11.46 min,5-乙基-2-甲基吡啶;8,12.86 min,四甲基吡嗪。
具体实施方式
下面结合实施例对本发明作进一步的详细描述,但其并不限制本发明。
本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过购买获得的常规产品。
本发明所述的萃取液层为萃取后萃取溶剂所在层。
本发明如无特殊说明,百分号代表质量百分数。
实施例1
一种分离测定烟草吸食者唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将0.2 g脱脂棉置于烟草吸食者口腔内,放置0.3 min后取出,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品。
步骤(2),萃取:移取1.0 mL唾液样品于离心管中,加入1.0 mL二氯甲烷,0.4 g氯化钠,于1000 rpm转速下涡旋振荡1.0 min,离心后取下层即二氯甲烷层,剩余上层液中加入1.0 mL二氯甲烷重复萃取依1次,离心后取下层即二氯甲烷层,合并两次萃取的二氯甲烷层即为萃取液;
步骤(3),测定:将步骤(2)收集到的萃取液于30℃下减压蒸发至1 mL,过微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,得到的样品的总离子流色谱图如图2所示,根据标准曲线计算烟草吸食者唾液中吡嗪类和吡啶类物质的含量。
气相色谱条件如下:色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;不分流进样;进样量:2 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃ /min 升温到280℃,保持5 min。
质谱条件如下:电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为180℃,电离能量为40 eV,溶剂延迟:4.0 min,反应气为甲烷,流量为40%,采集方式为总离子流色谱图(TIC)。
表2和表3是本方法分析烟草吸食者唾液中吡嗪类和吡啶类物质的试验数据。表2给出了本分析方法测定目标物特征离子的测定值,表明该方法适合于烟草吸食者唾液中吡嗪类和吡啶类物质的定性鉴定。表3给出了本分析方法的线性相关系数、精密度(RSD)、检出限、定量限和回收率相关参数,表明该方法定性准确,结果可靠,精密度好。此外,样品前处理过程简单,操作简便,无需特殊仪器和试剂,萃取时加入盐与不加入盐相比显著提高了目标分析物的萃取率,萃取率提高了10-30%,并保证了保证目标物的稳定性,目标物回收率高,回收率大于91.1%。该方法适合于烟草吸食者唾液中吡嗪类和吡啶类物质的分析检测。
表2 特征离子测定值
表3线性相关系数、精密度(RSD)、检出限、定量限和回收率
n表示实验重复次数。
实施例2
一种分离测定白酒饮用者唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将1.0 g脱脂棉置于白酒饮用者口腔内,放置4 min后取出,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品。
步骤(2),萃取:移取5.0 mL唾液样品于离心管中,加入5.0 mL乙腈和2.0 mL甲苯,2.5 g无水硫酸镁、0.5 g氯化钠、0.5 g柠檬酸钠、0.25 g柠檬酸氢二钠,于10000 rpm转速下涡旋振荡5.0 min,离心后取上层即乙腈和甲苯层;
步骤(3),测定:将步骤(2)收集到的萃取液,40℃下减压蒸发至1 mL,过微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,得到的样品的总离子流色谱图如图3所示,根据标准曲线计算白酒饮用者唾液中吡嗪类和吡啶类物质的含量。
气相色谱条件如下:色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;分流进样,分流比为15;进样量:1 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃ /min 升温到280℃,保持5 min。
质谱条件如下:电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为200℃,电离能量为60 eV,溶剂延迟:5.0 min,反应气为甲烷,流量为60%,采集方式为总离子流色谱图(TIC)。
表4和表5是本方法分析白酒饮用者唾液中吡嗪类和吡啶类物质的试验数据。表4给出了本分析方法测定目标物特征离子的测定值,表明该方法定性准确。表5给出了本分析方法的线性相关系数、精密度(RSD)、检出限、定量限和回收率相关参数,表明该方法结果可靠,精密度好。此外,萃取时加入盐与不加入盐相比显著提高了目标分析物的萃取率,萃取率提高了15-35%,并保证了保证目标物的稳定性,目标物回收率高,回收率大于88.1%。该方法适合于白酒饮用者唾液中吡嗪类和吡啶类物质的分析检测。
表4 特征离子测定值
表5线性相关系数、精密度(RSD)、检出限、定量限和回收率
n表示实验重复次数。
实施例3
一种分离测定唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将0.1g脱脂棉置于口腔内,放置0.2min后,取出吸收了唾液的脱脂棉,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品;
步骤(2),萃取:移取0.5mL唾液样品于离心管中,加入萃取溶剂、盐和除水剂,经涡旋振荡至混合均匀后离心,取萃取液层,得到萃取液;涡旋振荡转速为100 rpm,涡旋振荡时间为0.2min,离心转速不低于10000 rpm,离心时间不少于5 min;
所述的萃取溶剂为二氯甲烷和乙腈(二氯甲烷和乙腈的体积比为3:1),萃取溶剂加入量与唾液样品的体积比为0.2:1;所述的盐为柠檬酸钠和柠檬酸氢二钠(柠檬酸钠和柠檬酸氢二钠的质量比为1:1),盐的加入质量为唾液样品质量的25%;所述的除水剂为无水硫酸镁,除水剂的加入量为唾液样品质量的70%;
步骤(3),测定:将步骤(2)收集到的萃取液过0.22 μm微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,外标法定量;
其中,气相色谱条件如下:
色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱或相当者;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;分流进样,分流比1;进样量:1 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃/min 升温到280℃,保持5 min。
飞行时间质谱条件如下:
电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为160℃,电离能量为40 eV,溶剂延迟:4.0min,反应气为甲烷,流量为40%%,采集方式为总离子流色谱图(TIC),监测的特征离子参数见表1。
外标法定量的具体方法是:配制吡嗪类和吡啶类物质系列标准溶液,浓度为0.05 mg/L、0.1 mg/L、0.5 mg/L、1 mg/L、2 mg/L、5 mg/L、10 mg/L,对系列标准溶液进行气相色谱-飞行时间质谱联用仪分析,得到标准溶液的总离子流色谱图,根据各测定物质特征离子峰面积为纵坐标,以各测定物质浓度为横坐标,作各测定物质的标准曲线;根据标准曲线计算样品中吡嗪类和吡啶类物质含量,以两次平行测定的平均值为最终测定结果。所述的吡嗪类物质为2-甲基吡嗪、2,6-二甲基吡嗪、2,3-二甲基吡嗪、三甲基吡嗪和四甲基吡嗪;所述的吡啶类物质为吡啶、3-乙基吡啶和5-乙基-2-甲基吡啶。
实施例4
一种分离测定唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将2.0g脱脂棉置于口腔内,放置5 min后,取出吸收了唾液的脱脂棉,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品;
步骤(2),萃取:移取10 mL唾液样品于离心管中,加入萃取溶剂、盐和除水剂,经涡旋振荡至混合均匀后离心,取萃取液层,得到萃取液;取萃取液层后留下的溶液再加入萃取溶剂,重复与步骤(2)相同的萃取方式萃取,萃取后合并萃取液层;
重复萃取的次数为3次;
所述的萃取溶剂为三氯甲烷和乙酸乙酯(三氯甲烷和乙酸乙酯的体积比为1:1)。每次萃取溶剂的用量与唾液样品的体积比为2:1
涡旋振荡转速为20000 rpm,涡旋振荡时间为10 min,离心转速不低于10000 rpm,离心时间不少于5 min;
所述的盐为氯化钠和柠檬酸钠的组合物,质量比为1:0.5,盐的加入质量为唾液样品质量的10%;
所述的除水剂为无水硫酸钠,每次除水剂的加入质量为唾液样品质量的10%;
步骤(3),测定:将步骤(2)收集到的萃取液浓缩后过0.22 μm微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,外标法定量;
浓缩的方法为减压蒸发(压力没有限制),减压蒸发的温度为20℃,浓缩至2 mL;
其中,气相色谱条件如下:
色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱或相当者;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;分流进样,分流比30;进样量:1.2 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃/min 升温到280℃,保持5 min。
飞行时间质谱条件如下:
电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为220℃,电离能量为60 eV,溶剂延迟: 6.0 min,反应气为甲烷,流量为60%,采集方式为总离子流色谱图(TIC),监测的特征离子参数见表1。
外标法定量的具体方法是:配制吡嗪类和吡啶类物质系列标准溶液,浓度为0.05 mg/L、0.1 mg/L、0.5 mg/L、1 mg/L、2 mg/L、5 mg/L、10 mg/L,对系列标准溶液进行气相色谱-飞行时间质谱联用仪分析,得到标准溶液的总离子流色谱图,根据各测定物质特征离子峰面积为纵坐标,以各测定物质浓度为横坐标,作各测定物质的标准曲线;根据标准曲线计算样品中吡嗪类和吡啶类物质含量,以两次平行测定的平均值为最终测定结果。所述的吡嗪类物质为2-甲基吡嗪、2,6-二甲基吡嗪、2,3-二甲基吡嗪、三甲基吡嗪和四甲基吡嗪;所述的吡啶类物质为吡啶、3-乙基吡啶和5-乙基-2-甲基吡啶。
实施例5
一种分离测定唾液中吡嗪类和吡啶类物质的方法,包括以下步骤:
步骤(1),样品采集:将1g脱脂棉置于口腔内,放置3min后,取出吸收了唾液的脱脂棉,置于注射器针筒中,利用注射器推杆推移挤压,挤出液收集于样品管中,得到唾液样品;
步骤(2),萃取:移取5 mL唾液样品于离心管中,加入萃取溶剂和盐,经涡旋振荡至混合均匀后离心,取萃取液层,得到萃取液;取萃取液层后留下的溶液再加入萃取溶剂,重复与步骤(2)相同的萃取方式萃取,萃取后合并萃取液层;
重复萃取的次数为1次;
涡旋振荡转速为5000 rpm,涡旋振荡时间为5 min,离心转速不低于10000 rpm,离心时间不少于5 min;
所述的萃取溶剂为二氯甲烷、三氯甲烷、甲苯、乙酸乙酯和乙腈的组合物(二氯甲烷、三氯甲烷、甲苯、乙酸乙酯和乙腈的体积比为1:1:1:1:1),萃取溶剂加入量与唾液样品的体积比为1:1;
所述的盐为氯化钠柠檬酸钠和柠檬酸氢二钠的组合物,质量比为1:1:1,盐的加入质量为唾液样品质量的30%;
所述的除水剂为无水硫酸镁和无水硫酸钠的组合物,质量比为1:1,除水剂的加入量为唾液样品质量的50%;
步骤(3),测定:将步骤(2)收集到的萃取液浓缩后过0.22 μm微孔滤膜,滤液采用气相色谱-正化学源-飞行时间质谱法分析,外标法定量;
浓缩的方法为减压蒸发(压力没有限制),减压蒸发的温度为50℃,浓缩至1 mL;
其中,气相色谱条件如下:
色谱柱为长度×内径×膜厚为30 m×0.25 mm × 0.25 μm,DB-35MS石英毛细管柱或相当者;载气氦气,纯度≥99. 999 %,恒流模式,流量1.0 mL/min;进样口温度:280℃;不分流进样;进样量:1.5 μL;程序升温:初始温度35℃,保持 2 min,然后以7.5℃/min升温到125℃,再以25 ℃/min 升温到280℃,保持5 min。
飞行时间质谱条件如下:
电离方式为正化学电离源(PCI),色谱质谱接口温度为280℃,离子源温度为200℃,电离能量为50 eV,溶剂延迟:5.0 min,反应气为甲烷,流量为50%,采集方式为总离子流色谱图(TIC),监测的特征离子参数见表1。
外标法定量的具体方法是:配制吡嗪类和吡啶类物质系列标准溶液,浓度为0.05 mg/L、0.1 mg/L、0.5 mg/L、1 mg/L、2 mg/L、5 mg/L、10 mg/L,对系列标准溶液进行气相色谱-飞行时间质谱联用仪分析,得到标准溶液的总离子流色谱图,根据各测定物质特征离子峰面积为纵坐标,以各测定物质浓度为横坐标,作各测定物质的标准曲线;根据标准曲线计算样品中吡嗪类和吡啶类物质含量,以两次平行测定的平均值为最终测定结果。所述的吡嗪类物质为2-甲基吡嗪、2,6-二甲基吡嗪、2,3-二甲基吡嗪、三甲基吡嗪和四甲基吡嗪;所述的吡啶类物质为吡啶、3-乙基吡啶和5-乙基-2-甲基吡啶。
实施例6
实施例6与实施例5的区别在于:步骤(2)重复萃取的次数为2次;步骤(3)浓缩温度为40℃,浓缩至1.5 mL。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!