酮咯酸氨丁三醇或其制剂中酮咯酸氨丁三醇或/和杂质的HPLC检测方法与流程
本发明属于物质检测技术领域,特别是涉及酮咯酸氨丁三醇或其制剂中酮咯酸氨丁三醇或/和杂质的HPLC检测方法。
背景技术:
酮咯酸氨丁三醇,英文名称Ketorolac Tromethamine,简称KTT,CAS号74103-07-4。酮咯酸氨丁三醇是一种非甾体类抗炎药,能抑制前列腺素生物合成,生物活性与其S-型有关。动物研究显示酮咯酸氨丁三醇有镇痛作用,无镇静或抗焦虑作用。
根据KTT合成路线,可知其可能出现的杂质主要为降解杂质,比如,酮咯酸和氨丁三醇反应生成杂质A(CAS号167105-80-8),酮咯酸在合成过程被氧化生成杂质B(CAS号154476-25-2,1-羟基酮咯酸)、杂质C(CAS号113502-52-6,1-酮基酮咯酸)、杂质D和杂质E(CAS号80965-09-9),原料药中需对这5个降解杂质进行控制。另外,KTT注射液处方中由于有乙醇作为辅料,酸和醇可能发生反应生成酯,故制剂中除了控制原料药中的5个降解杂质,还需杂质F(CAS号108061-03-6)。
USP和EP药典中针对KTT原料药杂质的液相色谱检测中,存在以下不足:流动相盐相均为50mmol/L的磷酸二氢铵(用磷酸调节pH至3.0),有机相为四氢呋喃,而四氢呋喃会对检测仪器造成一定的伤害。
技术实现要素:
有鉴于此,本发明拟对KTT原料药及其制剂中杂质的HPLC检测方法进行优化改进,同时,本发明所述方法亦能够对KTT原料药及其制剂中KTT进行定性或定量检测。
本发明通过以下技术方案实现上述目的:
本发明提供了酮咯酸氨丁三醇或其制剂中杂质的检测方法,采用HPLC对待检溶液进行检测,根据色谱结果定性或定量,色谱条件包括:
色谱柱:C8硅胶色谱柱;
检测波长:检测波长200~400nm;
流动相及洗脱程序:流动相A为含有磷酸缓冲液的水相,pH为4.1~4.3;流动相B为乙腈和四氢呋喃的混合有机相,有机相中四氢呋喃的体积分数为10~15%;按如下梯度1~3任一程序进行梯度洗脱:
梯度1:
梯度2:
梯度3:
进一步,所述杂质包括杂质A、杂质B、杂质C、杂质D、杂质E、杂质F中的一种或两种以上的混合:
使用本发明检测方法,能够同时将上述A-F多个杂质与主成份进行有效分离,可以同时实现对其中一种或多种成分的有效检测。
进一步,所述磷酸缓冲液中含有磷酸二氢根离子和磷酸根离子,pH为4.1~4.3;进一步地,所述磷酸缓冲液为磷酸二氢铵-磷酸缓冲液;更进一步地,pH为4.2;更进一步地,磷酸二氢铵浓度为5.75g/L。
进一步,所述色谱柱规格为4.6×250mm,5μm。
进一步,流动相流速为0.6~1.2ml/min,进一步选自0.9~1.1ml/min。
进一步,柱温选自20~42℃,进一步选自38~42℃。
本发明中,柱温、流速、进样量等参数可以在常见范围内选择。
进一步,待检溶液的溶剂选自水或乙腈体积分数25%以下的乙腈水溶液;进一步地,所述待检溶液至少包括供试品溶液;更进一步地,待检溶液还包括对照品溶液。
本发明检测方法中,如果使用到对照品,则对照品与目标物质相对应。本发明中所述“对照品与目标物质相对应”,是指根据目标物质选取该化合物相对应的对照品,如目标物质为杂质A,则对应的就是杂质A对照品;若目标物质为杂质A和D,则对应的就是杂质A和D对照品。
本发明还提供了对酮咯酸氨丁三醇或其制剂中酮咯酸氨丁三醇的检测方法,采用HPLC对待检溶液进行检测,根据色谱结果定性或定量,色谱条件包括:
色谱柱:C8硅胶色谱柱;
检测波长:检测波长200~400nm;
流动相及洗脱程序:流动相A为含有磷酸缓冲液的水相,pH为4.1~4.3;流动相B为乙腈和四氢呋喃的混合有机相,有机相中四氢呋喃的体积分数为10~15%;按如下梯度1~3任一程序进行梯度洗脱:
梯度1:
梯度2:
梯度3:
本发明检测波长,可以在前述公开的范围内通过常规手段进行调节选择。在寻找最佳检测波长时,可以使用紫外分光光度法、HPLC配套使用的全波段扫描等方式进行,再配合HPLC检测器的检测效果(如避免溶剂干扰等),采用常规技术找到适宜的检测波长。本发明一个具体实施方式中,检测波长选自311~315nm,例如在313nm。
本发明研究表明,四氢呋喃的体积分数在10%时就能够达到本发明检测效果,实际操作中优先使用四氢呋喃含量低的流动相。
本发明中定性检测,可以使用常规方法进行,例如选用对照品通过外标法进行对应分析,又或者通过HPLC将各成分分开以后,通过常规鉴定手段进行定性分析,如质谱、薄层、紫外等等。
本发明中定量检测,可以使用外标法、面积归一法等常规方法进行含量计算。
另外,需要说明的是,本发明中的分离度、杂质限度的控制均通过本领域的常规技术手段进行调节,杂质限度可参照USP或EP药典。
在定量分析时,若使用外标法,采用常规手段制作标准曲线进行计算即可;但在定性分析时,则无需制作标准曲线,可以通过保留时间来判定。
本发明的有益效果是:本发明针对酮咯酸氨丁三醇原料药及其制剂尤其是注射液中相关杂质的HPLC检测方法进行优化改进,从检测波长、流动相体系的确定及供试品溶液浓度的筛选等角度进行优化和改进,并从系统适用性、降解专属性、检测限和定量限、线性及范围、精密度、准确度、溶液稳定性及方法耐用性等角度对本发明所述检测方法进行验证,结果显示,本发明所述方法具备良好的杂质检出能力,通过改进流动相,减少四氢呋喃的比例,既保证杂质的检出和分离,又减少四氢呋喃对柱子的损伤,另外本发明的梯度洗脱相对于等度洗脱来说,所用检测时间更短,该方法更加快捷简便,高效。
附图说明
图1KTT及各杂质的紫外吸收图谱,其中,a-g图依次为KTT、杂质A、杂质B、杂质C、杂质D、杂质E、杂质F的紫外吸收图谱;*DAD 1,22.252(60.6mAU,-)参考值=21.885&22.719为混合溶液1.D;
图2梯度1的典型液相色谱谱图;
图3梯度2的典型液相色谱谱图;
图4梯度3的典型液相色谱谱图;
图5梯度4的典型液相色谱谱图;
图6梯度5的典型液相色谱谱图;
图7梯度6的典型液相色谱谱图。
具体实施方式
本发明所用色谱仪器为Agilent 1260高效液相色谱仪。
酮咯酸氨丁三醇(KTT)及其注射液中杂质及各杂质的现有技术检测情况的控制限度见表1。
表1
其中,杂质A、B、C、D通过购买获得,杂质E(参照专利U.S.,5621115)和杂质F(参照“Kaneria,Ankur et al,Fe-EROS Encyclopedia of Reagents for Organic Synthesis,Triethyl Methanetricarboxylate,2001”)分别由实验室合成。
本发明所述酮咯酸氨丁三醇及其制剂尤其是注射液中杂质或/和酮咯酸氨丁三醇的HPLC检测方法,具体如下:
(一)色谱条件
色谱柱:ZORBAX Eclipse plus C8色谱柱,规格4.6×250mm,5μm,柱温40℃;
检测器:紫外检测器,检测波长313nm;
流动相及洗脱程序:5.75g/L磷酸二氢铵缓冲盐溶液用磷酸调pH值至4.2作为流动相A;乙腈:四氢呋喃=90:10(v/v)作为流动相B,按表2程序进行梯度洗脱,
表2
流速:1.0ml/min;
进样量:10μl。
(二)试剂配制
供试品溶液:精密量取供试品1ml(规格30mg)或2ml(规格15mg),置50ml量瓶中,用水稀释至刻度,摇匀,作为供试品溶液;每1ml中约含0.6mg的酮咯酸氨丁三醇;
对照品溶液:精密量取1ml对照品溶液,置于100ml量瓶中,用水稀释至刻度,摇匀,即得。
系统适用性溶液:精密称取杂质A、杂质B、杂质C、杂质D杂质E和杂质F 10mg,置10ml量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为混合杂质对照母液;精密量取0.6ml混合杂质对照母液,置10ml量瓶中,甲醇稀释至刻度,摇匀作为杂质对照品储备液;另精密称取酮咯酸氨丁三醇对照品60mg,置10ml量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为对照品储备液;精密量取杂质对照品储备液、对照品储备液各1ml,置10ml量瓶中,用水稀释至刻度,摇匀,制成每1ml中含杂质A、杂质B、杂质C、杂质D、杂质E、杂质F各约1.2μg,含酮咯酸氨丁三醇约0.6mg的混合溶液,作为系统适用性溶液。
(三)进样步骤
第一步,取水,进样1次;
第二步,取系统适用性溶液,进样1次;
第三步,取对照品溶液,进样1次;
第四步,取供试品溶液,进样1次。
(四)杂质含量计算
由以下公式计算:
其中:
rfi:为酮咯酸氨丁三醇各杂质峰面积的校正因子;
ri:为单个杂质的峰面积;
rs:为对照溶液的峰面积。
总杂质含量(%)=所有杂质含量之和。
各个条件的具体优化过程如下所述。
一、检测波长的确定
从KTT和杂质A、杂质B、杂质C、杂质D、杂质E、杂质F各自的定位图谱中提取对应的紫外吸收图谱,如图1所示,结果显示KTT在313nm处有最大吸收,杂质杂质A、杂质B、杂质C、杂质D、杂质E、杂质F均在313nm处有较大吸收,故选用313nm作为KTT原料药及注射液,包括杂质测定的检测波长。
二、流动相梯度确定
表3
以下实验均采用表3所述相关条件进行。
参考USP和EP药典,流动相盐相均为50mmol/L的磷酸二氢铵(用磷酸调节pH至3.0),有机相为四氢呋喃。考虑四氢呋喃对仪器的伤害较大,本发明尝试在有机相中添加乙腈降低替换四氢呋喃的比例。选用KTT混合溶液(包括KTT、杂质A、杂质B、杂质C、杂质D、杂质E和杂质F)进行流动相的筛选。表4为筛选的色谱条件1。
表4色谱条件筛选1
结果见图2,结果显示,系统适用性溶液杂质检出个数不足,可能有杂质峰相互重叠,且从主峰的峰形来看可能包裹有杂质,初步推测流动相pH对杂质的出峰时间有较大的影响。配制原条件流动相(以0.05mol/L的磷酸二氢铵缓冲液(用磷酸调节pH至3.0):四氢呋喃(70:30)),测得pH值为5.5,故下一步考察流动相A的pH值及计划采用梯度洗脱,调节流动相A的pH至与有机相混合在初始比例(A:B=80:20)下的pH为5.5左右,此时流动相A的pH约为4.2。表5为筛选流动相pH及梯度洗脱程序的色谱条件,分别进样混合溶液。
表5色谱条件筛选2
图3结果显示,混合溶液在梯度2色谱条件杂质全部检出,但是杂质C包裹有未知杂质,杂质E和相邻的未知杂质分离效果不佳;图4结果显示,混合溶液在梯度3色谱条件杂质全部检出,但是主峰包裹有未知杂质。故考虑改变主峰与杂质C附近的流动相比例,少量增加有机相中四氢呋喃的比例,考察主峰和杂质B、杂质C和未知杂质的分离效果是否可以改善。表6为筛选流动相梯度的色谱条件。分别在以下2个色谱条件下进样分析样品溶液。
表6色谱条件筛选3
结果见图5,样品溶液在梯度4的色谱条件下主峰的拖尾因子为0.9,选择梯度4的色谱条件对混合溶液和各杂质定位溶液进样分析,从混合溶液结果分析主峰的拖尾因子为0.9,各已知杂质完全分离,各已知杂质之间的分离度均大于3.0,杂质C与相邻未知杂质之间的分离度大于1.5,所有杂质在23min全部出峰,故考虑优化梯度后半段,缩短梯度时间。表7为对梯度4优化的色谱条件,对混合溶液进样分析。
表7色谱条件筛选4
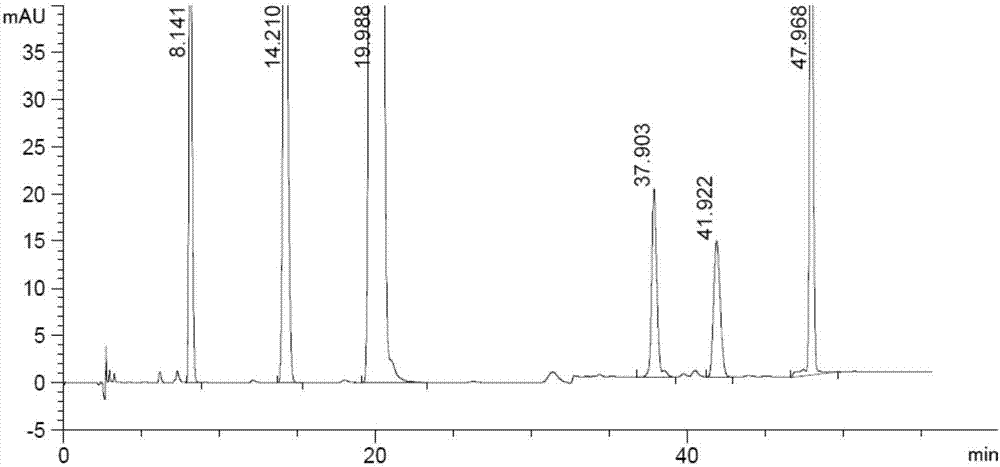
结果见图6,混合溶液在梯度5的色谱条件下主峰的拖尾因子为0.9,在0.8~1.5范围内,主峰与各已知杂质能达到基线分离要求,分离度均大于3.0;各已知杂质与其相邻的未知杂质的分离度均大于2.0,故此条件为最优条件。
鉴于梯度程序中15min至20min、20min至25min均为流动相A逐渐减少,故将梯度5的梯度程序优化。表8对梯度5优化的色谱条件,对混合溶液进样分析。
表8色谱条件筛选5
结果如图7所示,在优化后的色谱条件下进样分析混合溶液,主峰的拖尾因子在0.8~1.5范围内,主峰与各已知杂质能达到基线分离要求,分离度均大于3.0,故将此条件定为最终分析条件。
四、供试品溶液浓度的筛选
参考酮咯酸氨丁三醇USP40原料药的质量标准,考虑有关物质检测中对杂质的检测灵敏度,本发明将供试品溶液的浓度初步制定为0.6mg/ml,进样体积为10μl。试验结果表明:在优化的条件下对样品进行分析,主峰未过载,且拖尾因子在0.8~1.5范围内,理论版数大于5500,因此,将供试品溶液的浓度初步制定为0.6mg/ml,进样体积为10μl。
五、进样量的确定
供试品绝对进样量(6μg)低于USP注射液有关物质的绝对进样量(20μg),但高于USP38/40、BP2013、EP8.0的绝对进样量(4μg)。同时,在本发明选定的流动相pH值下,本品及各个已知杂质的紫外最大吸收均在313nm附近,主峰及已知杂质在313nm检测波长的响应值远高于药典规定的在254nm检测波长处的响应值。因此,并不会影响对杂质的检出能力。
采用本发明所述检测方法与USP药典的检测方法分别检测了中试6批成品的有关物质,将两个方法的结果对比,结果见表9和10,结果显示,本发明所述检测方法对杂质的检出能力不劣于USP注射液有关物质的检测方法。
表9注射液(15mg规格)两种方法对比检测结果
备注:检测样品为长期稳定性9月的样品。
表10注射液(30mg规格)两种方法对比检测结果
备注:检测样品为长期稳定性9月的样品。
进一步,本发明从系统适用性、降解专属性、检测限和定量限、线性及范围、精密度、准确度、溶液稳定性及方法耐用性等角度对所述检测方法进行验证,验证结果见表11。
表11
以上验证结果表明,本发明所述检测方法具备良好的杂质检出能力。
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!