UPLC法快速测定西红花有效成分含量及鉴别非法染色的制作方法
本发明属于药物分析化学领域,具体涉及西红花多种成分的检测方法。
背景技术:
西红花又称藏红花、番红花,为鸢尾科植物番红花crocussativusl.的干燥柱头,具有活血化瘀,凉血解毒,解郁安神之功效。西红花由于特殊的用药部位使得其产量低,价格昂贵,市场上造假、掺假的情况十分严重,原料质量参差不齐,非法染色情况甚是严峻。在对市场造成冲击的同时,其中的一些偶氮类色素对人体也存在一定的健康危害。而原料的好坏直接决定了产品的品质,所以有必要对西红花进行全面的质量控制,有效成分含量测定和非法染色鉴别缺一不可。
西红花苷(crocins)是其主要活性成分,西红花药材的质量优劣与西红花苷含量密切相关。为了加强监管,国家食品药品监督管理局发布了药品补充检验方法和检验项目批准件2011001,对金胺o、新品红、柠檬黄和胭脂红4种常见非法染色物进行检验。近年来,检测西红花药材质量及鉴别非法染色的文献报道也较多,但未见同时定量西红花苷-ⅰ、西红花苷-ⅱ和鉴别金胺o、新品红、柠檬黄和胭脂红4种常见染料的检测方法。两项检测工作分开进行,严重影响了工作效率,故有必要建立西红花高效、快速的整体表征和质量控制方法。
现有技术中检测西红花药材质量及鉴别非法染色的法规及文献报道较多。西红花品质评价分析方面,日本药典-jp16、英国药典-bp2013、欧洲药典-ep8及iso-3632都采用紫外-可见分光光度法定量西红花总苷来评价西红花的质量;
2015年版《中国药典》采用hplc法检测西红花苷ⅰ和西红花苷ⅱ含量以评价西红花的质量;也有文献报道采用近红外光谱及拉曼光谱技术均建立了较好的西红花苷定量模型];非法染色鉴别方面,张彬彬等建立了一种薄层色谱-表面增强拉曼光谱法成功检测经金胺o、新品红、柠檬黄和胭脂红4种常见染料染色的西红花药材。付凌燕等采用液-质联用以及对照试剂对照的方法,对市场上西红花非法添加的色素鉴定为柠檬黄、胭脂红、金胺o和新品红,又建立薄层色谱和高效液相色谱法鉴别非法染色情况。但未见同时定量西红花苷-ⅰ、西红花苷-ⅱ和检测金胺o、新品红、柠檬黄和胭脂红4种常见染料的检测方法。
技术实现要素:
本发明现将《中国药典》2015版中有效成分的检测方法同药典补充方法里对柠檬黄、胭脂红、金胺o和新品红4种目前市场上常见非法色素的分析方法结合起来,联合使用uplc定时波长自动切换技术及梯度洗脱法,快速测定西红花有效成分西红花苷-ⅰ、西红花苷-ⅱ含量及鉴别柠檬黄、胭脂红、金胺o和新品红4种常见非法染色物,方法方便快捷、准确度高,仪器设备要求低,大大提高工作效率的同时减少名贵药材的消耗及相关实验所带来的时间及经济成本,为企业产品的生产打下良好的基础。
首先,本发明提供了一种uplc法波长切换技术测定西红花有效成分含量及鉴别非法染色物的方法,其包括如下步骤:
(1)配制含有西红花苷-i、西红花苷-ⅱ、柠檬黄、金胺o、新品红和胭脂红的混合对照品溶液;
(2)将待测样品配制成西红花供试品溶液;
(3)取色谱柱,将步骤(1)所述混合对照品溶液和步骤(2)所述西红花供试品溶液分别上色谱柱,用洗脱液进行梯度洗脱;其中,所述洗脱液由流动相a和流动相b组成,流动相a为乙腈,流动相b为乙酸铵-乙酸水溶液;梯度洗脱中流动相a在洗脱液中所占体积分数为:
第0~3min,流动相a体积分数从10%匀速上升到37%;
第3~9min,流动相a体积分数=37%;
第9~10min,流动相a体积分数从37%匀速下降到10%;
(4)采用uplc定时波长自动切换技术,对不同时间段的组分测定不同波长下的吸光值,所述0min是从步骤(3)中进样开始计时:
第0~2.0min,在432nm波长下检测柠檬黄;
第2.0~4.0min,在509nm波长下检测胭脂红;
第4.0~5.3min,在440nm波长下检测西红花苷-i和西红花苷-ⅱ;
第5.3~6.5min,在550nm波长下检测新品红;
第6.5~7.0min,在432nm波长下检测金胺o;
第7.0~8.0min,在550nm波长下检测新品红;
第8.0~8.7min,在432nm波长下检测金胺o;
第8.7~10.0min,在550nm波长下检测新品红;
(5)将西红花供试品溶液的谱图和混合对照品溶液的谱图进行比对,对待测样品中的西红花苷-i、西红花苷-ⅱ、柠檬黄、金胺o、新品红和胭脂红进行定性检测;
(6)配制含有西红花苷-i、西红花苷-ⅱ的混合对照品,并稀释至系列质量浓度,然后按照步骤(3)和(4)的色谱条件上样测定;采用外标法进行定量;
其中,步骤(6)中的外标法优选为:以峰面积为纵坐标,对照品浓度为横坐标,绘制标准曲线,分别得到西红花苷-i、西红花苷-ⅱ的线性回归方程,将待测样品的峰面积代入线性回归方程,对待测样品中的西红花苷-i、西红花苷-ⅱ进行定量检测。
优选地,步骤(1)所述洗脱液的体积流速为0.1~0.5ml/min,更优选为0.3ml/min;
优选地,步骤(1)所述流动相b为含0.05%乙酸的0.05mol/l乙酸铵水溶液,即所述乙酸铵-乙酸水溶液中乙酸铵的摩尔浓度为0.05mol/l,乙酸的体积分数为0.05%,其配制方法可以为:取3.854g乙酸铵和0.5ml乙酸,用蒸馏水稀释至1l。
优选地,步骤(1)所述混合对照品溶液的配制方法可以为:分别精密称取西红花苷-i、西红花苷-ⅱ、柠檬黄、金胺o、新品红和胭脂红对照品适量,置于5ml容量瓶中,分别使用流动相a与流动相b体积比1:9的溶液将它们配制成质量浓度分别约为0.42、0.40、0.41、0.44、0.49、0.42mg/ml的对照品储备溶液;精密吸取上述对照品储备溶液各1ml于同一个10ml容量瓶中,加入体积分数70%的乙醇稀释定容,得混合对照品溶液;
优选地,步骤(1)中所述混合对照品溶液的上样量为0.1~2μl,更优选为1μl;
优选地,步骤(2)所述西红花供试品溶液的配制方法可以为:精密称取西红花药材粉末20~60mg(如40mg),加入乙醇50~90ml(如90ml),再以流动相a和流动相b体积比1:9的混合溶液定容至100ml,摇匀后,过滤溶液得到。
其中,所述乙醇优选为体积分数70%的乙醇;
进一步优选地,在加入乙醇后,对溶液进行超声处理15~30min(如20min),超声优选在冰浴条件下进行;
优选地,步骤(2)中所述西红花供试品溶液的上样量为0.1~2μl,更优选为1μl。
本发明实验过程中发现以乙醇(比如70%乙醇)溶液配置的柠檬黄对照品,在梯度洗脱中色谱峰出现分叉现象,原因推测为对照品溶剂与流动相极性相差较大,而使用流动相a和流动相b体积比1:9的混合溶液作为溶剂,色谱峰正常。故本发明在配制混合对照品溶液时,优选的溶剂为流动相a(乙腈)和流动相b(乙酸铵-乙酸水溶液)体积比1:9的混合溶液。
优选地,步骤(3)所述色谱柱为acquityuplchsst3色谱柱;柱温优选为32.5~37.5℃(如35℃)。
本发明步骤(3)中流动相a和流动相b的体积比通过watersempower软体建立曲线6模式,即匀速模式。
本发明所述的方法中,理论塔板数按所测各成分色谱峰计均不低于10000,分离度均不小于1.5。
本发明所述uplc法波长切换技术测定西红花有效成分含量及鉴别非法染色物的方法,可以对西红花有效成分(西红花苷-i、西红花苷-ⅱ)进行定性定量检测,同时可以对非法染色物(柠檬黄、金胺o、新品红和胭脂红)进行定性检测。
由于非法染色物的含量通常较低,因此,为了进一步保证测定方法的准确性,优选地,在前述uplc法波长切换技术测定西红花有效成分含量及鉴别非法染色物的方法中,还包括如下步骤:
(7)将西红花供试品溶液的色谱图和混合对照品溶液的色谱图进行比对后,若供试品溶液的色谱图中出现与对照品溶液的色谱图保留时间一致的谱峰,则对该谱峰进行光谱扫描(可以采用二极管阵列检测器全波长扫描);
(8)若该谱峰的光谱图与对照品的光谱图也一致,则比较它们的色谱纯度数据。
本发明利用uplc定时波长自动切换技术将西红花中的2种有效成分与4种常见非法染色物的特征峰同时体现在一张色谱图上,可更直观地判断样品中是否存在可疑色素及进行含量测定,避免了测定单个成分、逐步判断的繁琐步骤,准确灵敏、重复性好,且对仪器设备要求较低,在企业质检部门的实际工作中可操作性强,能帮助企业在短时间内较全面地评价西红花药材的质量,为企业原料质量控制提供参考。
本发明对色谱峰的确认采用保留时间、紫外光谱图、色谱峰纯度数据三者相结合的方法。样品检测时,若某个供试品的色谱图中出现与对照品保留时间一致的谱峰,对其进行光谱扫描,若该谱峰的光谱图与对照品一致,同时比较其色谱纯度数据,三者结合则可初步判断样品中是否检出该成分。
本发明鉴定的4种染色物中,柠檬黄、胭脂红为偶氮磺酸盐类色素,极性和水溶性较强,反相色谱保留较弱;而金胺o和新品红为碱性芳基甲烷类染料,反相色谱保留较强,极性差别较大。为建立同时分离4种染色物的检测方法,本发明先后试验筛选了3种色谱柱杂化颗粒(桥式乙基硅氧烷硅胶杂化颗粒、表面带电杂化颗粒、高强度硅胶颗粒),t3柱具有平衡保留极性与非极性分析物的优势,通过对3种杂化颗粒的分离效果、耐用性等因素综合考虑,最后选择acquityuplchsst3色谱柱,取得了良好的效果。
附图说明
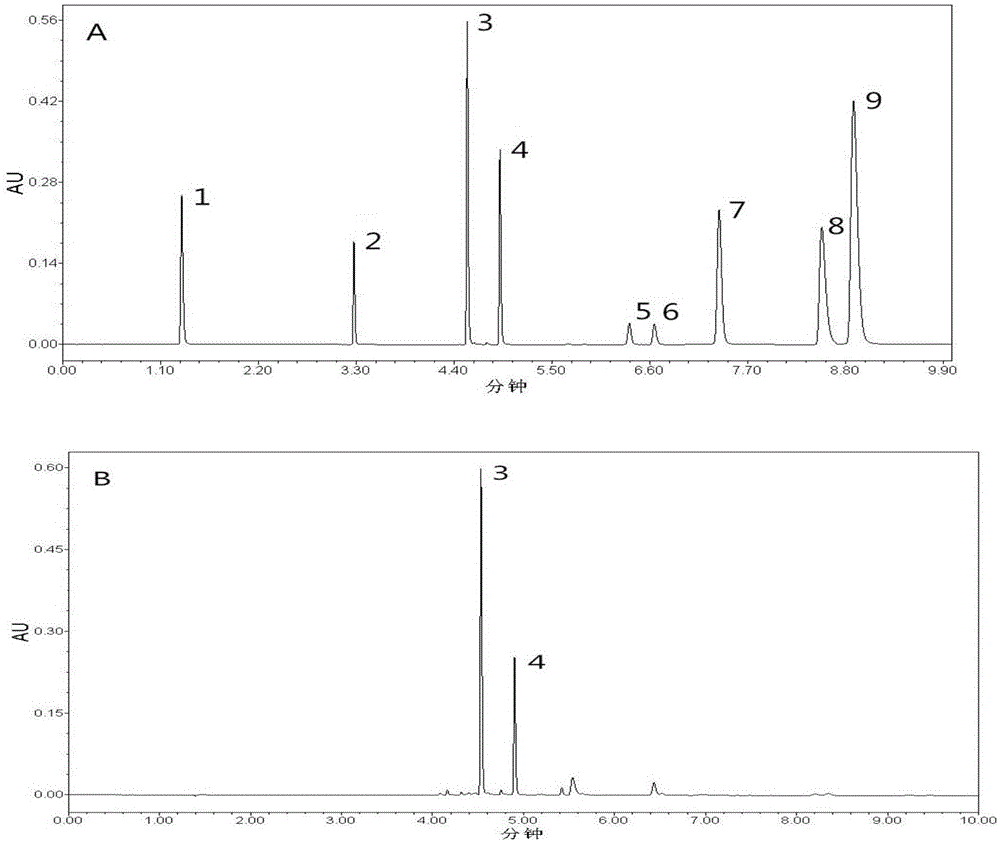
图1为实施例1中1号混合对照品溶液的高效液相色谱图(a)和编号s9供试品溶液的色谱图(b);9个峰分别为:1.柠檬黄;2.胭脂红;3.西红花苷-i;4.西红花苷-ⅱ;5、7和9.新品红;6和8.金胺o;
图2为待测化合物的紫外光谱图;9个图分别为:1.柠檬黄;2.胭脂红;3.
西红花苷-i;4.西红花苷-ⅱ;5、7和9.新品红;6和8.金胺o。
具体实施方式
仪器与材料
acquityuplch-classsystem超高效液相色谱系统;kq5200v型超声波清洗器(昆山市超声仪器有限公司);sartoriussqp电子分析天平(赛多利斯科学仪器有限公司);milli-qreference超纯水机(德国默克密理博公司);thermofisherpico17高速离心机(德国thermofisher公司)。
对照品西红花苷-i(批号111588-201704,含量≥88.4%)、西红花苷-ⅱ(批号111589-201705,含量≥92.2%)、柠檬黄(批号510004-201602,含量≥86.9%)、新品红(批号111955-201301,含量≥71.5%)、金胺o(批号111770-201603)、胭脂红(批号111771-201302)均购买于中国食品药品检定研究院。乙腈为色谱纯;乙酸为优级纯;乙酸铵、无水乙醇为分析纯;水为超纯水。
西红花样品购自于上海崇明岛、安徽亳州、浙江杭州、伊朗、青海西宁和西藏6个地区,共12批,样品信息见表1。经过上海医药集团股份有限公司中央研究院教授级工程师叶冠鉴定为鸢尾科植物番红花crocussativusl.的干燥柱头。
实施例中为流动相a为乙腈,流动相b为0.05mol/l乙酸铵水溶液(含0.05%乙酸),配制方法为:取3.854g乙酸铵和0.5ml乙酸,用蒸馏水稀释至1l。
实施例1
对照品溶液的制备
分别取西红花苷-i、西红花苷-ⅱ、柠檬黄、金胺o、新品红和胭脂红对照品适量,精密称定,置于5ml容量瓶中,分别使用流动相a与流动相b体积比1:9的溶液将它们配制成质量浓度分别约为0.42、0.40、0.41、0.44、0.49、0.42mg/ml的对照品储备溶液。精密吸取上述对照品储备溶液各1ml于同一个10ml容量瓶中,加入体积分数70%的乙醇稀释定容,得1号混合对照品溶液。
供试品溶液的制备
分别称取12批次的西红花药材粉末约40mg,精密称定,置于100ml棕色容量瓶中,加入体积分数70%乙醇溶液90ml,冰浴超声(功率200w,频率40khz)20min后放至室温,再以流动相a和流动相b体积比1:9混合溶液定容,摇匀,滤过,取滤液,即得编号s1-s12的西红花供试品溶液。
一种快速测定西红花有效成分含量及鉴别非法染色物的方法,其包括如下步骤:
(1)采用acquityuplchsst3(2.1mm×150mm,1.8μm)色谱柱;柱温35℃;将供试品溶液上色谱柱,上样量为1μl,用洗脱液进行梯度洗脱;其中,所述洗脱液由流动相a和流动相b组成,流动相a为乙腈,流动相b为乙酸铵-乙酸水溶液;梯度洗脱中流动相a在洗脱液中所占体积分数为:
第0~3min,流动相a体积分数从10%匀速上升到37%;
第3~9min,流动相a体积分数=37%;
第9~10min,流动相a体积分数从37%匀速下降到10%;
(2)对不同时间段的组分测定不同波长下的吸光值:
第0~2.0min,在432nm波长下检测柠檬黄;
第2.0~4.0min,在509nm波长下检测胭脂红;
第4.0~5.3min,在440nm波长下检测红花苷-i和西红花苷-ⅱ;
第5.3~6.5min,在550nm波长下检测新品红;
第6.5~7.0min,在432nm波长下检测金胺o;
第7.0~8.0min,在550nm波长下检测新品红;
第8.0~8.7min,在432nm波长下检测金胺o;
第8.7~10.0min,在550nm波长下检测新品红。
理论塔板数按所测各成分色谱峰计均不低于10000,分离度均不小于1.5。
依照前述同样的方法,将1号混合对照品溶液上色谱柱,得到1号混合对照品溶液的色谱图,见图1(a)。
分别精密吸取前述配制的西红花苷-i和西红花苷-ⅱ储备溶液各0.5ml于1ml棕色容量瓶中,得2号混合对照品溶液。再用体积分数70%乙醇稀释至10、20、50、100、200、500、1000倍,得到系列质量浓度的混合对照品溶液,按实施例1中的色谱条件分别进行上样进样测定。以峰面积为纵坐标(y),对照品浓度c(mg/ml)为横坐标(x),绘制标准曲线。得西红花苷-i的线性范围0.042~0.00042mg/ml,回归方程y=1.89×107x-3240,r=0.9999;西红花苷-ⅱ的线性范围0.040~0.00040mg/ml,回归方程y=2.61×107x-4210,r=0.9999。
将编号s1-s12的西红花供试品溶液的色谱图与1号混合对照品溶液色谱图比对,对西红花有效成分(西红花苷-i、西红花苷-ⅱ)和非法染色物(柠檬黄、金胺o、新品红和胭脂红)进行定性检测(其中,图1(b)为编号s9供试品溶液的色谱图)。将编号s1-s12的西红花供试品溶液的西红花苷-i、西红花苷-ⅱ的峰面积代入西红花苷-i、西红花苷-ⅱ的线性回归方程,对西红花苷-i、西红花苷-ⅱ进行定量检测。12批西红花样品的测定结果见表1。
表1样品信息与测定结果
本品按《中国药典》西红花干燥品含西红花苷-i(c44h64o24)和西红花苷-ⅱ(c38h54o19)的总量不得少于10.0%的要求,对照实验中所测得样品结果发现,12批样品中有9批符合要求,另3批有效成分含量明显偏低(s8、s11、s12)。通过对样品中可疑峰与4种色素对照品的紫外光谱图对照及峰纯度检测后发现s8、s11、s12号样品存在染色现象,分别检出新品红、柠檬黄、金胺o和胭脂红,按照药典补充规定应不得检出非法染色物。
为进一步保证结果的准确性,本发明对色谱峰的确认还采用了色谱图保留时间、紫外光谱图、色谱峰纯度数据三者相结合的方法。样品检测时,若某个供试品的色谱图中出现与对照品色谱图保留时间一致的谱峰,则对其进行光谱扫描,若该谱峰的光谱图与对照品一致,同时比较其色谱纯度数据,三者结合则可初步判断样品中是否检出该成分。对照品的光谱图如图2所示。进一步的检测结果显示与表1中的结果相一致
实施例2检测方法的验证
2.1有效成分含量测定
2.1.1精密度
取实施例1中制备得到的编号s9供试品溶液,按实施例1中的色谱条件分别进行上样进样测定,分别于1天内重复进样6次和连续3天内重复进样3次,每次进样1μl,测定西红花苷-i、西红花苷-ⅱ的峰面积,计算日内及日间精密度。日内精密度评价中,西红花苷-i、西红花苷-ⅱ峰面积的rsd分别为0.99%、0.43%。日间精密度评价中,西红花苷-i、西红花苷-ⅱ峰面积的rsd分别为1.06%、1.05%。
2.1.2稳定性试验
取实施例1中制备得到的编号s9供试品溶液,分别于配制后在室温下放置0、2、4、6、8、12、24h,按实施例1中的色谱条件分别进行上样进样测定,得西红花苷-i、西红花苷-ⅱ峰面积的rsd分别为1.40%和1.21%,表明供试品溶液制备后24h内稳定。
2.1.3重复性
按照实施例1中供试品溶液的配制方法,取s9批次的西红花样品平行制备供试品溶液6份,分别按实施例1中的色谱条件分别进行上样进样测定,结果西红花苷-i、西红花苷-ⅱ质量分数的rsd值分别为1.30%、1.70%。
2.1.4耐用性
通过改变柱温(32.5、35、37.5℃)、和流动相ph(5.3、5.5、5.7),其余按照实施例1中的色谱条件分别进行上样进样测定。结果表明,色谱条件作微小改变,其分离效果未发生明显变化,各条件下西红花苷-i、西红花苷-ⅱ峰面积的rsd均<3%,该方法耐用性良好。
2.1.5加样回收率
取已知含量的同一批(s9批次)西红花粉末6份,每份约40mg,精密称定,置100ml容量瓶中,分别精密加入相当于样品成分量100%的各对照品溶液,按实施例1中所述方法制备供试品溶液,按实施例1中的色谱条件分别进行上样进样测定。结果西红花苷-i、西红花苷-ⅱ的平均回收率分别为100.14%、101.64%,rsd分别为2.04%、2.21%。
2.2非法染色鉴别——检测限与定量限
将实施例1中配制的柠檬黄、金胺o、新品红、胭脂红储备溶液逐级稀释,按实施例1中的色谱条件分别进行上样进样测定。结果当信噪比(s/n)=3时,柠檬黄、金胺o、新品红、胭脂红的检测限分别为0.041、0.088、0.11、0.14ng;当s/n=10时,定量限分别0.17、0.23、0.32、0.42ng。
- 还没有人留言评论。精彩留言会获得点赞!