一种用制备色谱分离复杂物中目标物的优化方法与流程
本发明涉及色谱技术领域,尤其涉及一种用制备色谱分离复杂物中目标物的优化方法。
背景技术:
制备色谱法(通常指色谱单柱)是分离复杂混合物中目标组分的重要手段,为提高目标组分的纯度、收率、产率、生产效率及减小溶剂消耗,对色谱分离条件进行优化是必要的。通过测定吸附等温线建立被分离组分的色谱模型,进行计算优化,不仅能够快速找到优化点,还可以省去烦琐的实验。
吸附等温线反映了色谱柱中固定相对流动相中溶质i的吸附能力,表示为单位质量的固定相上吸附溶质的量qi(x,t)与流动相中溶质浓度ci(x,t)的关系,即qi(x,t)=f(c1(x,t),c2(x,t),...,ci(x,t),...),吸附等温线有多种表示形式,如:langmuir、bilangmuir、freundlich和tóth【林炳昌.模拟移动床色谱技术.化学工业出版社,北京:2008】,这里所涉及到的各组分的初始浓度必须已知,才可能有明确的吸附等温线。迄今为止,所有两组分及多组分色谱模型都是针对相关组分浓度已知的混合物的分离。用色谱法从复杂混合物中,如:天然产物,提纯目标组分时,经常遇到干扰组分浓度未知的情况,所以无法获得吸附等温线,也就无法获得被分离组分的色谱模型,此时的优化过程建立在大量的实验基础上。
技术实现要素:
本发明的目的是针对现有制备色谱进行复杂混合物分离时操作参数优化方法技术的不足,提供一种用制备色谱分离复杂物中目标物的优化方法。
本发明的具体技术方案如下:
一种用制备色谱分离复杂混合物中目标物的优化方法,包括以下步骤:
1、配制复杂混合物的进样液,复杂混合物含有3种以上组分,并测定其中目标物的浓度;
2、确定在制备色谱所用固定相和流动相体系中目标物的干扰组分,并赋予干扰组分的浓度;
3、用进样液采集2-5条目标物及其干扰组分的实际制备色谱的流出曲线数据;
4、选择单一组分的吸附等温线描述目标物的吸附行为;选择单一组分的吸附等温线描述干扰组分的吸附行为;
5、选择色谱模型分别描述目标物和干扰组分的色谱行为;再用差分法求解目标物和干扰组分的色谱模型,进而分别得到单柱目标物和干扰组分的计算流出曲线;
6、根据实际流出曲线用逆法确定:目标物的吸附等温线参数和色谱模型的计算参数;干扰组分的吸附等温线参数和色谱模型的计算参数;
7、用色谱模型优化制备色谱分离目标物和干扰组分的操作条件;
8、在由色谱模型优化出来的分离条件下进行实际分离。
上述方法中,当所述干扰组分结构未知,并且其浓度无法测定时,假定干扰组分浓度。
上述方法中,所述色谱模型为理想色谱模型。
上述方法中,所述理想色谱模型的差分求解方法如下:
qi=f(ci)(2)
式(1)为理想色谱模型,式(2)为吸附等温线,其中i代表组分i,i=1为目标物,i=2为干扰组分;ci,qi分别为组分在流动相与固定相的浓度;u为流动相的流速;f为相比,f=(1-ε)/ε,ε为孔隙率;t代表时间,x代表位置;
进而,将式(1)写成
式中,
再将式(3)中
式中,τ为时间步长,h为空间步长,j表示当前的塔板,j=1,2,...,l/h,n代表时间,n=kτ,k=1,2,3...,显式差分格式稳定性条件:
或者将式(3)中
上述的两种差分格式边界条件:
通过对上述显式差分格式或者隐式差分格式迭代求解,获得组分i流出曲线ci(l,t)-t,l为色谱柱长。
上述方法中,所述的根据实际流出曲线用逆法确定吸附等温线参数、模型计算参数h和τ时,需要经过设定初值并经模型求解至计算流出曲线与实际流出曲线吻合后确定最终取值;
所述的使计算流出曲线与实际流出曲线吻合的方法为,利用色谱模型计算色谱流出曲线,并和实际测定的流出曲线进行比较,按下式计算两者误差平方和:
上述方法中,所述色谱模型优化制备色谱分离目标物和干扰组分的操作条件是按单因素法或单纯形或正交设计或均匀设计或响应面设计或遗传算法进行的。
上述方法中,按制备色谱的流动相的配比配制复杂混合物的进样液。
上述方法中,所述的采集目标物及其干扰组分的实际制备色谱的流出曲线数据为采集线性条件下目标物及其干扰组分的实际制备色谱的流出曲线数据,以及采集过载条件下目标物及其干扰组分的实际制备色谱的流出曲线数据。
上述方法中,干扰组分结构未知的情况下,假定进样液仅含目标组分1和干扰组分2,计算结果以目标物的提纯指标为约束条件和优化目标函数,以流动相流速、原料进样浓度、原料进样时间、截取目标物流出液时间段为优化参数。
具体地说,混合液是复杂混合物的溶液,至少为3组分,假定混合液仅含目标组分1和干扰组分2,用制备色谱分离时,干扰组分2流出曲线与目标组分1流出曲线相邻,干扰组分2是所有杂质中严重干扰目标组分1分离的杂质;计算结果以目标物的提纯指标为约束条件和优化目标函数,如:计算结果需满足目标物的纯度约束条件:≥95%,以生产效率或者收率为目标函数;再如:计算结果需满足目标物的收率,以生产效率或者纯度或者生产效率和纯度为目标函数;
优化问题描述:
max:p=f(c1,f,tp,u,t)
其中,c1,f为进料液目标组分浓度,tp为进样时间,t为截取目标物流出液的时间段,t=tend-tstart,tstart为截取目标物馏分开始时间,tend为截取目标物馏分结束时间,u为流动相线速度(
目标物纯度pu1计算公式:
目标物收率y1的计算公式:
色谱分离的生产率的计算公式:
本发明的有益效果是:
1、简化建模过程
本发明在复杂混合物色谱分离的体系中,仅对目标物和干扰组分用理想色谱模型建模,色谱分离过程的吸附、脱附、传质、扩散、竞争均通过逆法求解得到的单组份吸附等温线的参数和理想色谱模型算法的参数来体现,用最简单的但却是最主要的形式处理最为复杂的而且是无法解决的建模问题。而在现有的复杂色谱体系中,即使所有组分已知,传质系数、扩散系数、竞争关系也无法准确确定,也就无法准确建模。
2、模型求解方法简单准确
本发明采用差分格式迭代求解,方程求解误差小,在线性及非线性体系下具有良好的稳定性,目标物的计算色谱流出曲线与实验流出曲线吻合良好,而且假定浓度的干扰组分的计算色谱流出曲线与实验流出曲线吻合也较好。
3、用模型指导复杂混合物色谱分离实践
本发明对于所用的复杂混合物色谱分离的体系,确定复杂混合物进样液中与目标物相邻的干扰组分后,干扰组分结构未知、浓度无法测定时,或者根据进样液中目标物浓度、干扰组分检测信号和目标物检测信号赋予干扰组分的浓度,或者根据进样液中目标物浓度、进样液在制备色谱的干扰组分流出曲线色谱峰面积和目标物流出曲线色谱峰面积赋予干扰组分的浓度,该杂质浓度为假定浓度,由此通过逆法获得假定的吸附等温线并建立假定的理想色谱模型,可以粗略预测干扰组分的色谱行为。对于未知杂质,虽然没有相应的标准物质,由实验测得的未知杂质流出曲线无法用浓度和时间表示,只能用响应信号和时间表示,但是该流出曲线能反映出与各组分的分离情况,当赋予未知杂质假定浓度后,其色谱峰峰底宽度和峰高位置依然准确,仍然能反映出与其它组分的分离程度,可以建模粗略描述其色谱行为。按目标组分1和干扰组分2考虑,当要求的分离后目标物纯度高于95%时,在模型获取的优化条件下分离,实际得到的纯度接近或高于90%(本专利中纯度均指hplc纯度)。
具体的,利用本发明的方法分离丹参中丹参酮iia时,按目标组分1和干扰组分2考虑,模型预测分离结果是:丹参酮iia的纯度为95.15%,收率89.19%,生产率2.35g/(h·l);实际分离结果是:丹参酮iia的纯度为93.15%,收率82.42%,生产率2.23g/(h·l),与模型预测分离结果吻合。按全组分考虑,通过这种优化法将丹参酮iia的纯度由38.89%提高至93.15%,丹参酮iia的收率82.42%,生产率2.23g/(h·l)。
利用本发明的方法分离喜树果中喜果苷时,按目标组分1和干扰组分2考虑,模型预测分离结果是:喜果苷纯度为95.28%,收率90.34%,生产率0.221g/h/l;实际分离结果是:喜果苷纯度为94.58%,收率91.7%,生产率0.224g/h/l,与模型预测分离结果吻合。按全组分考虑,通过这种优化法将喜果苷的纯度由25.78%提高至73.89%,喜果苷的收率91.7%,生产率0.224g/(h·l)。
利用本发明的方法分离甜叶菊中莱鲍迪a苷时,按目标组分1和干扰组分2考虑,模型预测分离结果是:莱鲍迪a苷的收率为99.09%,纯度97.86%;实际分离结果是:莱鲍迪a苷的收率为97.31%,纯度95.98%,与模型预测分离结果吻合。按全组分考虑,通过这种优化法将莱鲍迪a苷的纯度由8.73%提高至67.72%,莱鲍迪a苷的收率95.98%。
4、实验点少
选择吸附等温线后,需要2-5条实际流出曲线即可用逆法建模。如:在langmuir吸附等温线
5、本发明能够通过模型模拟获得色谱分离复杂混合物的优化操作条件,这是一种省时、省力、省原料和流动相的制备色谱寻优方法。
附图说明
图1是实施例1中色谱分离丹参中丹参酮iia的流出曲线图谱;
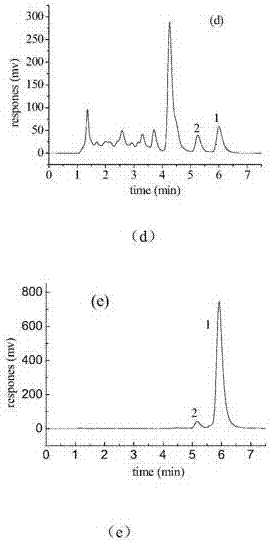
其中,(a)进样液hplc图谱,(b1)制备柱的第一条实际流出曲线和模拟流出曲线,(b2)制备柱的第二条实际流出曲线和模拟流出曲线,(c)经色谱模型优化的分离条件下的实际流出曲线和模拟流出曲线,(d)经色谱模型优化分离后干扰组分馏分的hplc谱图,(e)经色谱模型优化分离后目标物馏分的hplc谱图;虚线为模型计算得到的模拟流出曲线,实线为测得的实际流出曲线;1为目标物,2为干扰组分;
图2是实施例2中色谱分离喜树果提取物中喜果苷的流出曲线图谱;
说明如图1;
图3是实施例3中色谱分离甜叶菊提取物中莱鲍迪a苷的流出曲线图谱;
其中,(b3)制备柱的第三条实际流出曲线,其他说明如图1。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。
实施例1
一种用制备色谱从丹参萃取液中分离丹参酮iia的优化方法。
所用制备色谱体系:制备色谱柱:d×l=10mm×100mm;固定相:c18,20μm;流动相:乙醇-水,配比65∶35;色谱分离温度:室温;hplc检测条件:4.6mm×150mm分析柱(c18,5μm,agilent,u.s.a.),流动相甲醇与水的配比为85/15(v/v),流速1.0ml/min,测定波长270nm,测定温度30℃。
优化方法包括以下步骤:
(1)按制备色谱的流动相乙醇与水的配比配制丹参的进样液,用hplc测定丹参进样液中丹参酮iia的浓度,hplc谱图(见图1(a))中显示丹参酮iia(峰1)的纯度38.89%;
(2)通过制备色谱的流出曲线,确定进样液在该制备色谱体系中目标物1的干扰组分2,用hplc测得的进样液中干扰组分2检测信号与目标物丹参酮iia检测信号的比值k再与目标物浓度c1,f乘积,赋予干扰组分的浓度,该干扰组分浓度为假定浓度,c2,f=kc1,f,这里,k=0.15,c2,f=0.032mg/ml;
(3)测定线性条件下和过载条件下的制备色谱的2条实际流出曲线,分别为图1(b1),图1(b2);
第1条实际流出曲线的色谱条件:
进样液:丹参酮iia的浓度c1,f=0.22mg/ml
流速q:1ml/min
进样时间:5s
在线获得实际流出曲线;
第2条实际流出曲线的色谱条件:
进样液:丹参酮iia的浓度c1,f=0.22mg/ml
流速q:3ml/min
进样时间:2min
离线hplc检测;
(4)选择langmuir吸附等温线
(5)用理想色谱模型方程(1)分别描述目标组分1和前干扰组分2的色谱行为;再用显示差分法将理想色谱模型表示为:
(6)根据实际流出曲线用逆法确定:目标物1的吸附等温线参数g1、b1和色谱模型的计算参数τ、h;干扰组分的吸附等温线参数g2、b2;
根据在线性条件下获得的第1条实际流出曲线(图1(b1))和公式tri=t0(1+fgi),式中,tri为保留时间,t0为死时间,f为相比,获得吸附系数gi,丹参酮iia的吸附参数g1=10.75,其前干扰组分g2=7.89;
根据在进样量过载的非线性条件下获得的第2条实际流出曲线(图1(b2)),测定吸附系数bi,并确定h/τ;具体的,首先为参数赋初值,利用色谱模型计算色谱流出曲线,并和实际测定的曲线进行比较,按下式计算两者误差平方和:
(7)用色谱模型优化制备色谱分离目标物和干扰组分的操作条件:
假定丹参进样液仅含丹参酮iia和前干扰组分,计算结果需满足丹参酮iia的纯度约束条件:≥95%;
优化问题描述:
max目标物生产率:p=f(c1,f,tp,u,tstart)
决策变量和固定参数:
0.12mg/ml≤c1,f≤0.24mg/ml
2min≤tp≤6min
5.17cm/min≤u≤8.17cm/min
10min≤tstart≤15min
按单因素法或单纯形或正交设计或均匀设计或响应面设计或遗传算法进行色谱模型优化制备色谱分离丹参酮iia和前干扰组分的操作条件。这里采用遗传算法(ga)进行优化,ga参数为:种群规模:20,染色体长度:45bits,交叉概率:0.9,变异概率:0.1,进化代数:50;
结合ga,由模型优化的色谱分离条件为:c1,f=0.24mg/ml,c2,f=0.034mg/ml,tp=3.86min,u=7.39cm/min,tstart=10.48min,该条件下的预测分离结果是:得到丹参酮iia的纯度为95.15%,收率89.19%,生产率2.35g/(h·l);
(8)在由色谱模型优化出来的分离条件进行实际分离:
测定在色谱模型优化的分离条件下的实际流出曲线,为图1(c),0-10.48min杂质馏分和10.48-15min目标物馏分的hplc色谱图见图1(d)和图1(e),图中表明丹参酮iia和前干扰组分得到较好分离。实际得到丹参酮iia的纯度为93.15%,收率82.42%,生产率2.23g/(h·l),优化点的模型模拟结果与实验结果比较吻合。
实施例2
一种用制备色谱从喜树果萃取液中分离喜果苷的优化方法。
所用制备色谱体系:制备色谱柱:d×l=20mm×500mm;固定相:c18,20μm;流动相:乙醇-水,配比60∶40;色谱分离温度:室温。hplc检测条件:4.6mm×250mm分析柱(c18,5μm,agilent,u.s.a.),流动相甲醇与水的配比为45/55(v/v),流速1.0ml/min,测定波长256nm,测定温度25℃。
优化方法包括以下步骤:
(1)按制备色谱的流动相乙醇与水的配比配制喜树果的进样液,用hplc测定喜树果进样液中喜果苷的浓度,hplc谱图(图2(a))中显示喜果苷组分纯度16.90%;
(2)通过制备色谱的流出曲线,确定进样液在该制备色谱体系中目标物1的干扰组分2,根据进样液在制备色谱的干扰组分流出曲线色谱峰面积和目标物流出曲线色谱峰面积赋予干扰组分的浓度,该干扰组分浓度为假定浓度,c2f=c1,f;
(3)测定线性条件下的制备色谱的实际流出曲线,为图2(b1),测定过载条件下制备色谱的实际流出曲线,为图2(b2);
第1条实际流出曲线的色谱条件:
流速q:11.4ml/min
进样液:喜果苷的浓度为0.27mg/ml
进样时间:3s
在线获得实际流出曲线;
第2条实际流出曲线的色谱条件:
流速q:11.4ml/min
进样液:喜果苷的浓度为0.27mg/ml
进样时间:1min
离线hplc检测;
(4)同实施例1步骤(4);
(5)同实施例1步骤(5);
(6)按实施例1步骤(6),根据在线性条件下获得的第1条实际流出曲线(图2(b1))和公式tri=t0(1+fgi),测定吸附系数gi,可得喜果苷的吸附参数g1=1.36,其前干扰组分g2=0.4;根据在进样量过载的非线性条件下获得的喜果苷的第二条实际流出曲线(图2(b2)),测定喜果苷的b1=0.1,并设定前干扰组分b2=0,τ1=τ2=0.05,h1=h2=0.02。理想色谱模型模拟计算的流出曲线(虚线)与实验测定的流出曲线(实线)吻合较好;
(7)按实施例1步骤(7)用色谱模型优化制备色谱分离喜果苷和干扰组分的操作条件:
假定喜果进样液仅含喜果苷和前干扰组分,计算结果需满足喜果苷的纯度约束条件:≥95%;
优化问题描述:
max目标物生产率:p=f(c1,f,tp,u,tstart)
决策变量和固定参数:
0.09mg/ml≤c1,f≤0.3mg/ml
2min≤tp≤10min
4cm/min≤u≤8cm/min
10min≤tstart≤20min
由模型优化的色谱分离条件为:c1,f=c2,f=0.3mg/ml,tp=4.563min,u=5.66cm/min,tstart=15.02min,该条件下的预测分离结果是:得到喜果苷的纯度为95.28%,收率90.349%,生产率0.221g/(h·l);
(8)在由模型优化出来的色谱分离条件进行实际分离:
测定在模型优化的色谱分离条件下的实际流出曲线,为图2(c),13-14.5min的杂质馏分和15-20.5min的目标物馏分的hplc图谱见图2(d)和图2(e),表明喜果苷和前干扰组分得到较好分离。按目标组分1和干扰组分2考虑,实际得到喜果苷的纯度为纯度为94.58%,收率91.7%,生产率0.224g/(h·l),优化点的模型模拟结果与实验结果较好吻合;按全组分考虑,实际目标物纯度为73.89%,收率91.7%,生产率0.224g/(h·l)。
实施例3
一种用制备色谱从甜叶菊萃取液中分离莱鲍迪苷a(ra)的优化方法。
所用制备色谱体系:制备色谱柱:d×l=50mm×100mm;固定相:c18,40~60μm;流动相:乙醇-水,配比1∶1;色谱分离温度:室温。hplc检测条件:分析柱:4.6mm×250mm(c18,5μm,agilent,u.s.a.),流动相:色谱乙腈与ph=2.6的磷酸二氢钠缓冲液配比为32/68(v/v),流速1.0ml/min,测定波长213nm,测定温度30℃。
优化方法包括以下步骤:
(1)按制备色谱的流动相乙醇与水的配比配制甜叶菊的进样液,用hplc测定甜叶菊进样液中ra的浓度c1,f,hplc谱图3(a)显示ra纯度8.73%;
(2)通过制备色谱的流出曲线,确定进样液在该制备色谱体系中与目标物1相邻的干扰组分即未知杂质2,用步骤(1)hplc测得的进样液中干扰组分检测信号与目标物检测信号的比值k再与目标物浓度c1,f的乘积,赋予干扰组分的浓度,该干扰组分浓度为假定浓度,c2,f=kc1,f,这里,k=2;
(3)分别测定1条线性条件下制备色谱实际流出曲线(见图3(b1))和2条过载条件下的制备色谱实际流出曲线,分别见图3(b2)和图3(b3);
第1条实际流出曲线的色谱条件:
进样液中ra的浓度c1,f=25mg/ml
流速q:15ml/min
进样时间:0.5min
离线hplc检测;
第2条实际流出曲线的色谱条件:
进样液中ra的浓度c1,f=20mg/ml
流速q:20ml/min
进样时间:3min
离线hplc检测;
第3条实际流出曲线的色谱条件:
进样液中ra的浓度c1,f=25mg/ml
流速q:30ml/min
进样时间:2min
离线hplc检测;
(4)同实施例1步骤(4);
(5)同实施例1步骤(5);
(6)按实施例1步骤(6),根据线性条件下获得的第1条实际流出曲线(图3(b1))和公式tri=t0(1+fgi),可得ra的吸附参数g1=4.56,其前干扰组分g2=2.31;根据在进样量过载的非线性条件下获得的第2条(图3(b2))和第3条实际流出曲线和(图3(b3)),测定ra的b1=0.02,h1=0.02,τ1=0.25;前干扰组分b2=0.002,h2=0.1,τ2=0.25。理想色谱模型模拟计算的流出曲线(虚线)与实验测定的流出曲线(实线)吻合较好;
(7)用色谱模型优化制备色谱分离目标物和干扰组分的操作条件:
假定甜叶菊进样液仅含ra和前干扰组分,计算结果需满足ra的收率约束条件:≥99%;
优化问题描述:
max目标物纯度和生产率:p=f(c1,f,tp,u,tstart)=pc×puθ,式中θ为偏重指数因子,这里θ=20。
决策变量和固定参数:
5mg/ml≤c1,f≤25mg/ml
1min≤tp≤5min
12.5ml/min≤q≤40ml/min
5min≤tstart≤35min
结合ga,由模型优化的分离条件为:c1,f=13.8mg/ml,tp=2.16min,q=14.9ml/min,tstart=24.77min,该条件下的预测分离结果是:得到ra的收率为99.09%,纯度为97.86%;
(8)在色谱模型优化的分离条件进行实际分离:
测定在色谱模型优化的分离条件下的实际流出曲线,为图3(c),1-25min杂质馏分和26-36min目标物馏分的hplc色谱图分别见图3(d)和图3(e),由此表明ra和前干扰组分得到较好分离。按目标组分1(ra)和干扰组分2考虑,实际得到ra的收率为97.31%,纯度95.98%,优化点的模型模拟结果与实验结果吻合;按全组分考虑,实际ra纯度为67.72%,收率95.98%。
- 还没有人留言评论。精彩留言会获得点赞!