正极活性物质前体及其制备方法、正极活性物质及其制备方法以及包括所述正极活性物质的锂二次电池与流程
本发明涉及一种正极活性物质前体及其制备方法、正极活性物质及其制备方法以及包括所述正极活性物质的锂二次电池。
背景技术:
锂二次电池的一个组成要素正极活性物质不仅直接贡献于电池能量密度的表现,还成为决定寿命特性等的主要因素。
与此相关,正在蓬勃开展针对所谓NCM(镍钴锰酸锂)等层状结构的镍系锂金属氧化物的研究,这是因为镍(Ni)含量越大越能表现高容量。
不过,若将镍系锂金属氧化物内镍的摩尔含量增加到80%以上,则具有其结构变得不稳定的问题。为了解决这种问题,已知有掺杂、涂布、形成浓度梯度等方法。
但是,对于具有不稳定结构的镍系锂金属氧化物颗粒,均匀地掺杂其内部或均匀地涂布其表面是非常困难的事情。而且,对于具有浓度梯度的镍系锂金属氧化物颗粒来说,已知根据其颗粒大小,镍的平均摩尔含量变得不同。
另外,人们还尝试将颗粒大小不同的两种正极活性物质用一定的比例混合,以提高正极的能量密度。这是所谓的双模(bi-modal)技术,该技术由粒径小的颗粒(以下,称为“小粒径颗粒”)填充粒径大的颗粒(以下,称为“大粒径颗粒”)之间的孔隙,从而在单位体积内聚集大量的正极活性物质。
为了实现这种双模技术,首先需要制备大粒径颗粒及小粒径颗粒。但是,已知通常5μm以上的大粒径颗粒由于其颗粒粒径大,难以均匀地控制内部组成,进而难以均匀地控制按颗粒大小的镍的平均摩尔含量、浓度梯度的形态、掺杂及表面涂层等。
技术实现要素:
为了解决上述问题,对于具有如下的核-壳浓度梯度(Core-Shell Gradient,CSG)的镍系金属氢氧化物颗粒,提出用于实现大粒径活性物质的最小核部半径,所述核-壳浓度梯度在核部中镍浓度保持恒定且在壳部中镍浓度急剧下降。
本发明的一个实施方案提供一种锂二次电池用正极活性物质前体,其包括镍系金属氢氧化物颗粒,所述镍系金属氢氧化物颗粒包括:作为镍的摩尔含量恒定区域的核部;及壳部,围绕所述核部的外表面,并在从与所述核部的边界面至最外廓的方向上具有镍的摩尔含量逐渐减少的浓度梯度(gradient),所述镍系金属氢氧化物颗粒的下述式1的值为50%以上。
[式1]R1/(Ri+D1)×100%
在所述式1中,R1为所述镍系金属氢氧化物颗粒内的核部的半径,D1为所述镍系金属氢氧化物颗粒内的壳部的厚度。
具体地,所述式1的值可以为75%以上。
另外,所述镍系金属氢氧化物颗粒的平均组成可由下述化学式1表示。
[化学式1]Ni1-w1-x1-y1-z1Cow1M1x1M2y1M3z1(OH)2-p1Xp1
在所述化学式1中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w1、x1、y1、z1及p1为分别满足0<w1≤0.2、0<x1≤0.2、0≤y1≤0.1、0≤z1≤0.1、0<w1+x1+y1+z1≤0.4、0≤p1≤0.1的值。
此时,所述镍系金属氢氧化物颗粒内的核部的组成在整个区域中可由下述化学式2表示。
[化学式2]Ni1-w2-x2-y2-z2Cow2M1x2M2y2M3z2(OH)2-p2Xp2
在所述化学式2中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w2、x2、y2、z2及p2为分别满足0≤w2≤0.1、0≤x2≤0.1、0≤y2≤0.1、0≤z2≤0.1、0≤w2+x2+y2+z2≤0.2、0≤p2≤0.05的值。
而且,所述镍系金属氢氧化物颗粒内的壳部的组成在所述边界面中可由所述化学式2表示,在所述最外廓中可由下述化学式3表示,从所述边界面到所述最外廓,镍(Ni)、M1、M2及M3各自的摩尔含量可以逐渐变化。
[化学式3]Ni1-w3-x3-y3-z3Cow2M1x3M2y3M3z3(OH)2-p3Xp3
在所述化学式3中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w3、x3、y3、z3及p3为分别满足0<w3≤0.3、0<x3≤0.3、0≤y3≤0.1、0≤z3≤0.1、0<w3+x3+y3+z3≤0.5、0≤p3≤0.1的值。
所述镍系金属氢氧化物颗粒的直径可以为10μm至30μm。
本发明的另一个实施方案提供一种锂二次电池用正极活性物质前体的制备方法,该方法包括:分别制备第一金属盐水溶液及第二金属盐水溶液的步骤,所述第一金属盐水溶液及第二金属盐水溶液包括镍原料物质、异种金属原料物质及水且所述镍原料物质的摩尔浓度彼此不同;第一共沉淀步骤,以恒定的速度向保持恒定的pH且被供应螯合剂的反应器中供应所述第一金属盐水溶液以形成核部;第二共沉淀步骤,在所述第一共沉淀步骤之后,逐渐减少所述第一金属盐水溶液的供应速度的同时,逐渐增加所述第二金属盐水溶液的供应速度,以形成围绕所述核部的外表面的壳部;所述方法在所述第二共沉淀步骤中获取由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,并控制所述第一共沉淀步骤及所述第二共沉淀步骤,以使对所述获取的镍系金属氢氧化物颗粒的下述式1的值成为50%以上。
[式1]R1/(R1+D1)×100%
在所述式1中,R1为所述镍系金属氢氧化物颗粒内的核部的半径,D1为所述镍系金属氢氧化物颗粒内的壳部的厚度。
具体地,所述第一共沉淀步骤及所述第二共沉淀步骤可控制为满足下述式2。
[式2]log(T1)/log(T1+T2)≈R1/(R1+D1)
在所述式2中,T1为第一共沉淀步骤的执行时间,T2为所述第二共沉淀步骤的执行时间。
更为具体地,所述第一共沉淀步骤可执行10小时以上。由此,能够沉淀从中心具有R1半径且镍的摩尔含量在整个区域中恒定的颗粒。
而且,在所述第二共沉淀步骤中,将在所述第一共沉淀步骤中沉淀的颗粒作为核部来形成壳部,由此能够获取由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,所述壳部为从所述核部的表面具有R2厚度且从与所述核部的表面相接的边界沿着厚度方向镍的摩尔含量逐渐减少的区域。
本发明的另一个实施方案提供一种锂二次电池用正极活性物质,其包括镍系锂金属氧化物颗粒,所述镍系锂金属氧化物颗粒包括:作为镍的摩尔含量恒定区域的核部;及壳部,围绕所述核部的外表面,并在从与所述核部的边界面至最外廓的方向上具有镍的摩尔含量逐渐减少的浓度梯度(gradient),所述镍系锂金属氧化物颗粒的下述式3的值为50%以上。
[式3]R2/(R2+D2)×100%
在所述式3中,R2为所述镍系金属氧化物颗粒内的核部的半径,D2为所述镍系金属氧化物颗粒内的壳部的厚度。
具体地,所述镍系锂金属氧化物颗粒的平均组成可由下述化学式4表示。
[化学式4]Li1+m[Ni1-w4-x4-y4-z4Cow4M1x4M2y4M3z4]1-mO2-p4Xp4
在所述化学式4中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w4、x4、y4、z4及p4为满足0<w4≤0.2、0<x4≤0.2、0≤y4≤0.2、0≤z4≤0.1、0<w4+x4+y4+z4≤0.4、0≤p4≤0.1的值,m为满足-0.05≤m≤0.25的值。
此时,所述镍系锂金属氧化物颗粒内的核部的组成在整个区域中可由下述化学式5表示。
[化学式5]Li1+m[Ni1-w5-x5-y5-z5Cow5M1x5M2y5M3z5]1-mO2-p5Xp5
在所述化学式5中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w5、x5、y5、z5及p5为分别满足0≤w5≤0.1、0≤x5≤0.1、0≤y5≤0.1、0≤z5≤0.1、0≤w5+x5+y5+z5≤0.2、0≤p5≤0.1的值,m为满足-0.05=m≤0.25的值。
而且,所述镍系锂金属氧化物颗粒内的壳部的组成在所述边界面可由所述化学式5表示,在所述最外廓可由下述化学式6表示,且从所述边界面到所述最外廓,镍(Ni)、M1、M2及M3各自的摩尔含量可以逐渐变化。
[化学式6]Lil+m[Ni1-w6-x6-y6-z6Cow6M1x6M2y6M3z6]1-mO2-p6Xp6
在所述化学式6中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w6、x6、y6、z6及p6为满足0<w6≤0.3、0<x6≤0.3、0≤y6≤0.1、0≤z6≤0.1、0<w6+x6+y6+z6≤0.5、0≤p6≤0.1的值,m为满足-0.05≤m≤0.25的值。
更为具体地,所述式2的值可以为75%以上。
另外,所述正极活性物质可进一步包括涂层,所述涂层围绕所述壳部的外表面并包括B、Mg、Zr、Al、Mn、Co或其组合的元素、所述元素的氧化物、非晶质化合物、锂离子导电性氧化物(例如,硼酸锂(Lithium borate)、硼硅酸锂(1ithium borosilicate))、高分子等。
本发明的又一个实施方案提供一种锂二次电池用正极活性物质的制备方法,该方法包括:分别制备第一金属盐水溶液及第二金属盐水溶液的步骤,所述第一金属盐水溶液及第二金属盐水溶液包括镍原料物质、异种金属原料物质及水且所述镍原料物质的摩尔浓度彼此不同;第一共沉淀步骤,以恒定的速度向保持恒定的pH且被供应螯合剂的反应器中供应所述第一金属盐水溶液以形成核部;及第二共沉淀步骤,在所述第一共沉淀步骤之后,逐渐减少所述第一金属盐水溶液的供应速度的同时,逐渐增加所述第二金属盐水溶液供应速度,以形成围绕所述核部的外表面的壳部;所述方法在所述第二共沉淀步骤中获取由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,并控制所述第一共沉淀步骤及所述第二共沉淀步骤,以使对所述获取的镍系金属氢氧化物颗粒的下述式1的值成为50%以上,在所述第二共沉淀步骤之后,包括烧成所述镍系金属氢氧化物颗粒及锂原料物质的混合物以获得镍系锂金属氧化物颗粒的步骤。
[式1]R1/(R1+D1)×100%
在所述式1中,R1为所述镍系金属氢氧化物颗粒内的核部的半径,D1为所述镍系金属氢氧化物颗粒内的壳部的厚度。
具体地,所述异种金属原料物质可以为Co原料物质、Mn原料物质、Al原料物质、Mg原料物质、Zr原料物质、Sn原料物质、Ca原料物质、Ge原料物质、Ga原料物质、B原料物质、Ti原料物质、Mo原料物质、Nb原料物质及W原料物质中的一种或其中两种以上物质的混合物。
在获取所述镍系锂金属氧化物颗粒的步骤之后,可进一步包括形成围绕所述镍系锂金属氧化物颗粒的外表面的涂层的步骤。
具体地,形成围绕所述镍系锂金属氧化物颗粒的外表面的涂层的步骤可包括:混合所述镍系锂金属氧化物颗粒及涂层原料物质的步骤;及热处理所述镍系锂金属氧化物颗粒及涂层原料物质的混合物的步骤。
本发明的再一个实施方案提供一种锂二次电池,该电池包括:含有上述锂二次电池用正极活性物质的正极;负极;及电解质。
本发明对具有CSG型浓度梯度的镍系金属氢氧化物颗粒提出用于实现大粒径活性物质的最小核部半径,从而能够均匀地控制按其颗粒大小的内部组成(镍的平均摩尔含量、浓度梯度的形态等)。
附图说明
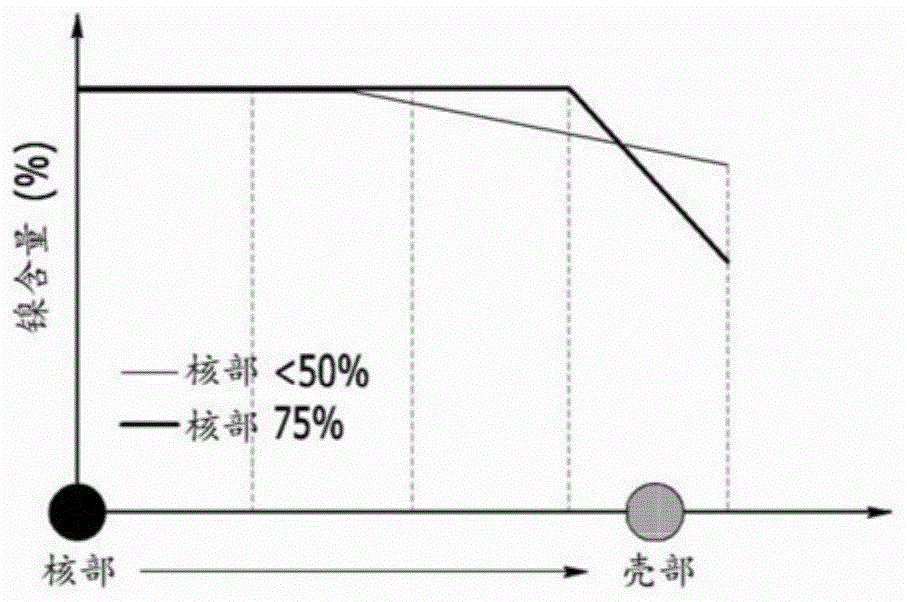
图1是示意地表示实施例1及比较例2各自的Ni含量分布的图。
图2a及图2b分别为对实施例2及比较例2各自的正极活性物质进行分级前(标为“参考”)的粒度分析(PSA,particle size analyze)结果。
图3a至图3e分别为在实施例2中分级的按直径的SEM照片(其中,图3a至图3e中的比例尺为50μm)。
图4a至图4d分别为在比较例2中分级的按直径的SEM照片(其中,图4a至图4d中的比例尺为20μm)。
图5a是表示对实施例2中分级前(标为“参考”)及分级后的各种正极活性物质进行的DSC(差示扫描量热法)热特性检测结果的图。
图5b是表示对比较例2中分级前(标为“参考”)及分级后的各种正极活性物质进行的DSC热特性检测结果的图。
图6是表示对实施例2及比较例2的分级前正极活性物质进行的直流内阻(DC-IR)增加率测量结果的图。
具体实施方式
下面,详细说明本发明的实施方案。但是,这仅是作为示例提出的,本发明并不限定于此,本发明仅由后述的权利要求书的范畴来定义。
此外,在本说明书中颗粒大小D0.9是指将分布有0.1、0.2、0.3…3、5、7……10、20、30μm这样多种颗粒大小的活性物质颗粒按体积比将颗粒累积至0.9%时的颗粒大小,D10是指按体积比将颗粒累积至10%时的颗粒大小,D50是指按体积比将颗粒累积至50%时的颗粒大小,D6是指按体积比将颗粒累积至6%时的颗粒大小,D95是指按体积比将颗粒累积至95%时的颗粒大小。
正极活性物质前体
本发明的一个实施方案提供一种锂二次电池用正极活性物质前体,其包括镍系金属氢氧化物颗粒,所述镍系金属氢氧化物颗粒包括:作为镍的摩尔含量恒定区域的核部;及壳体,围绕所述核部的外表面,并在从与所述核部的边界面至最外廓的方向上具有镍的摩尔含量逐渐减少的浓度梯度(gradient),所述镍系金属氢氧化物颗粒的下述式1的值为50%以上。
[式1]R1/(R1+D1)×100%
在所述式1中,R1为所述镍系金属氢氧化物颗粒内的核部的半径,D1为所述镍系金属氢氧化物颗粒内的壳部的厚度。
本发明的一个实施方案中提供的正极活性物质前体包括镍系金属氢氧化物颗粒,该颗粒具有如下的核-壳浓度梯度(Core-Shell Gradient,下称“CSG”),所述核-壳浓度梯度在核部中镍恒定地保持高浓度,在壳部中镍的浓度急剧下降。具体地,所述CSG形态有助于在核部中保持较高的镍的摩尔含量且高容量化,并且减少壳部中镍的摩尔含量并增加异种金属(例如,Mn、Co、Al等)的摩尔含量,从而将结构稳定化。
2)进一步,本发明的一个实施方案对所述具有CSG形态的镍系金属氢氧化物颗粒,通过所述式1提出用于实现大粒径活性物质的最小核部半径,从而能够均匀地控制按其颗粒大小的内部组成(镍的平均摩尔含量、浓度梯度的形态等)。
通常,具有浓度梯度的镍系金属氢氧化物颗粒是利用批处理(batch)式反应器,用共沉淀法制备。
据此,与利用连续搅拌槽反应器(Continuous Stirred Tank Reactor,CSTR)的情况相比,能够制备具有狭窄的高斯(Gaussian)分布的多元(plural)镍系金属氢氧化物颗粒。
尽管如此,使用所述批处理(batch)式反应器制成的具有浓度梯度的多元(plural)镍系金属氢氧化物颗粒存在着按其大小内部组成(镍的平均摩尔含量、浓度梯度的形态等)不同的问题。
这种问题在制备大粒径镍系金属氢氧化物颗粒时变得更加严重,基于此的大粒径活性物质的按颗粒大小的内部组成变得不均匀,最终制约双模活性物质的实现。
更为具体地,若以恒定的速度向保持恒定的pH并被供应螯合剂的批处理(batch)式反应器中供应所述第一金属盐水溶液,则能形成所述核部,将其随意称为“第一共沉淀步骤”。在所述第一共沉淀步骤之后,可连续地逐渐减少所述第一金属盐水溶液的供应速度的同时逐渐增加所述第二金属盐水溶液的供应速度,从而在所述核部的表面形成所述壳部。将其随意称为“第二共沉淀步骤”,可获取颗粒直径呈现高斯(Gaussian)分布的多元(plural)镍系金属氢氧化物颗粒,各种颗粒可具有CSG形态。
通常,已知将所述第一共沉淀步骤执行5小时左右,将所述第一共沉淀步骤及所述第二共沉淀步骤总共执行20小时左右来获取所述式1的值小于50%的所述多元(plural)镍系金属氢氧化物颗粒。此时,如上指出,由于金属盐水溶液的供应槽串联,存在着按其大小的内部组成(镍的平均摩尔含量、浓度梯度的形态等)变得不同的问题。
与此相反,如后述本发明的另一个实施方案,若适当地调节所述第一共沉淀步骤的执行时间和所述第一共沉淀步骤及所述第二共沉淀步骤的总执行时间,则能获取所述式1的值为50%以上的多元(plural)镍系金属氢氧化物颗粒。此时,即使利用串联的供应糟制备大粒径镍系金属氢氧化物颗粒,也能够使按照大小的内部组成(镍的平均摩尔含量、浓度梯度的形态等)变得均匀。具体地,在本发明的一个实施方案中所述式1的值可以为60%以上、或70%以上例如像后述实施例那样为75%以上。
对于详细的工序将在后面描述,下面详细说明包括所述镍系金属氢氧化物颗粒的正极活性物质前体。
镍系金属氢氧化物颗粒的大小
为了实现大粒径活性物质及基于此的双模活性物质,所述镍系金属氢氧化物颗粒可以是D50粒径为10μm至30μm的大粒径活性物质前体颗粒。
镍系金属氢氧化物颗粒的内部组成
所述镍系金属氢氧化物颗粒按其颗粒大小例如按D50粒径可具有由下述化学式1表示的平均组成。
[化学式1]Ni1-w1-x1-y1-z1Cow1M1x1M2y1M3z1(OH)2-p1Xp1
在所述化学式1中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w1、x1、y1、z1及p1为分别满足0<w1≤0.2、0<x1≤0.2、0≤y1≤0.1、0≤z1≤0.1、0<w1+x1+y1+z1≤0.4、0≤p1≤0.1的值。
这表示在由所述核部及所述壳部构成的镍系金属氢氧化物颗粒的整个区域中镍的平均摩尔含量为60%以上,是高浓度。
例如,在所述化学式1中,M1可以为Mn,0<w1≤0.1,0<x1≤0.1,y1=z1=p1=0,0<w1+x1+y1+z1≤0.2。
更为具体地,所述镍系金属氢氧化物颗粒按其颗粒大小例如按D50粒径可具有由下述化学式2表示的核部组成。
[化学式2]Ni1-w2-x2-y2-z2Cow2M1x2M2y2M3z2(OH)2-p2Xp2
在所述化学式2中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w2、x2、y2、z2及p2为分别满足0≤w2≤0.1、0≤x2≤0.1、0≤y2≤0.1、0≤z2≤0.1、0≤w2+x2+y2+z2≤0.2、0≤p2≤0.05的值。
这表示在所述镍系金属氢氧化物颗粒内的核部的整个区域中镍的摩尔含量为80%以上,其水平高于所述平均组成。
例如,在所述化学式2中,M1可以为Mn,0<w2≤0.05,0<x2≤0.05,y2=z2=p2=0,0<w2+x2+y2+z2≤0.1。
而且,所述镍系金属氢氧化物颗粒按其颗粒大小例如按D50粒径,在与所述核部的边界面上可由所述化学式2表示,在所述最外廓上可由下述化学式3表示,从所述边界面到所述最外廓,镍(Ni)、M1、M2及M3各自的摩尔含量可以逐渐变化。
[化学式3]Ni1-w3-x3-y3-z3Cow3M1x3M2y3M3z3(OH)2-p3Xp3
在所述化学式3中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w3、x3、y3、z3及p3为满足0<w3≤0.3、0<x3≤0.3、0≤y3≤0.1、0≤z3≤0.1、0<w3+x3+y3+z3≤0.5、0≤p3≤0.1的值。
这表示在所述镍系金属氢氧化物颗粒内与核部的边界面上镍的摩尔含量为80%以上,在所述最外廓上镍的摩尔含量为60%以上,具有从所述边界面到所述最外廓镍的摩尔含量逐渐减少的浓度梯度。与此同时,除了所述镍之外的金属(即,M1、M2及M3)具有从所述边界面到所述最外廓的摩尔含量逐渐增加的浓度梯度。
例如,在所述化学式3中,M1可以为Mn,0<w3≤0.25,0<x3≤0.25,y3=z3=p3=0,0<w3+x3+y3+z3≤0.5。
所述镍系金属氢氧化物颗粒根据所述化学式1的整体组成及所述化学式2的核部组成能够表现高容量的同时,根据所述化学式3的壳部组成能够具有稳定的结构。
正极活性物质前体的制备方法
本发明的另一个实施方案提供一种锂二次电池用正极活性物质前体的制备方法,该方法包括:分别制备第一金属盐水溶液及第二金属盐水溶液的步骤,所述第一金属盐水溶液及第二金属盐水溶液包括镍原料物质、异种金属原料物质及水且所述镍原料物质的摩尔含量彼此不同;第一共沉淀步骤,以恒定的速度向保持恒定的pH且被供应螯合剂的反应器中供应所述第一金属盐水溶液以形成核部;第二共沉淀步骤,在所述第一共沉淀步骤之后,逐渐减少所述第一金属盐水溶液的供应速度的同时,逐渐增加所述第二金属盐水溶液的供应速度,以形成围绕所述核部的外表面的壳部;所述方法在所述第二共沉淀步骤中获取由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,并控制所述第一共沉淀步骤及所述第二共沉淀步骤,以使对所述获取的镍系金属氢氧化物颗粒的下述式1的值为50%以上。
[式1]R1/(R1+D1)×100%
在所述式1中,R1为所述镍系金属氢氧化物颗粒内的核部的半径,D1为所述镍系金属氢氧化物颗粒内的壳部的厚度。
具体地,所述第一共沉淀步骤的执行时间可以与所述核部的半径R1成比例,所述壳部的形成时间可以与所述壳部的厚度D1成比例。
与此相关,所述第一共沉淀步骤及所述第二共沉淀步骤可控制为满足下述式2。
[式2]log(T1)/log(T1+T2)≈R1/(R1+D1)
在所述式2中,T1为第一共沉淀步骤的执行时间,T2为所述第二共沉淀步骤的执行时间。
下面,除去与上述说明重复的说明,说明所述各步骤的工序特征。
第一共沉淀步骤及第二共沉淀步骤的执行时间
所述第一共沉淀步骤的执行时间及所述第二共沉淀步骤的执行时间可适当地控制为所述式1的值成为50%以上,具体成为60%以上或70%以上,例如像后述实施例那样成为75%以上。
第一共沉淀步骤(核部形成步骤)
具体地,所述第一共沉淀步骤可执行10小时以上。由此,能够沉淀从中心的半径为R1且镍的摩尔含量在整个区域中恒定的颗粒。
这是通常将核部形成时间调节为5小时左右的大约两倍时间。将所述第一共沉淀步骤执行10小时以上,以形成具有长半径的核部,并执行满足所述式2的第二共沉淀步骤,因此有利于制备均匀地控制按其颗粒大小的内部组成(镍的平均摩尔含量、浓度梯度的形态等)的大粒径活性物质。
另外,在所述第一共沉淀步骤中使用的第一金属盐水溶液可以是以满足上述化学式2的化学计量摩尔比的方式混合镍原料物质、异种金属原料物质及水的水溶液。
所述镍原料物质为镍阳离子及任一阴离子离子结合的物质,凡是能够溶于水而解离成阳离子及阴离子的物质,并不特别限定。
而且,所述异种金属原料物质为除镍之外金属的阳离子(例如,Mn、Co、Al等的阳离子)及任一阴离子离子结合的物质,凡是能够溶于水而解离成阳离子及阴离子的物质,并不特别限定。例如,可以为Co原料物质、Mn原料物质、Al原料物质、Mg原料物质、Zr原料物质、Sn原料物质、Ca原料物质、Ge原料物质、Ga原料物质、B原料物质、Ti原料物质、Mo原料物质、Nb原料物质及W原料物质中的一种或其中两种以上物质的混合物。
第二共沉淀步骤(壳部形成步骤)
而且,在所述第二共沉淀步骤中,将在所述第一共沉淀步骤中沉淀的颗粒作为核部来形成壳部,由此能够获得由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,所述壳部为从所述核部的表面具有D1厚度且从与所述核部表面相接的边界面沿着厚度方向镍的摩尔含量逐渐减少的区域。
所述第二共沉淀步骤的执行时间可以考虑所述第一共沉淀步骤的执行时间和所述式2的值来确定。
在所述第一共沉淀步骤中使用的第二金属盐水溶液可以为,以满足上述化学式3的化学计量摩尔比的方式混合镍原料物质、异种金属原料物质及水的水溶液。
对于所述镍原料物质及异种金属原料物质的说明如上所述。
正极活性物质
本发明的另一个实施方案提供一种锂二次电池用正极活性物质,其包括镍系锂金属氧化物颗粒,所述镍系锂金属氧化物颗粒包括:作为镍的摩尔含量恒定区域的核部;及壳部,围绕所述核部的外表面,并在从与所述核部的边界面至最外廓的方向上具有镍的摩尔含量逐渐减少的浓度梯度(gradient),所述镍系锂金属氧化物颗粒的下述式3的值为50%以上。
[式3]R2/(R2+D2)×100%
在所述式3中,R2为所述镍系金属氧化物颗粒内的核部的半径,D2为所述镍系金属氧化物颗粒内的壳部的厚度。
本发明一个实施方案的正极活性物质可在如上所述正极活性物质前体的基础上制备。因此,关于所述正极活性物质的CSG形态、核部的半径及壳部的厚度关系(即,所述式2)等的说明与上述正极活性物质前体的说明相应。
具体地,所述正极活性物质是混合上述正极活性物质前体和锂原料物质,并且根据情况添加掺杂原料物质混合之后经过烧成而获取的物质,可包括多元(plural)镍系锂金属氧化物颗粒。
由此,与作为原料使用的正极活性物质前体同样地,1)具有CSG形态,即在核部中镍保持恒定的高浓度,在壳部中镍的浓度急剧下降;2)所述式2的值可以为50%以上、60%以上或70%以上,例如像后述实施例那样可以为75%以上。
对于具体的工序将在后面描述,下面详细说明包括所述镍系锂金属水化物颗粒的正极活性物质。
镍系锂金属氧化物颗粒的大小及高斯分布
所述镍系锂金属氧化物颗粒可以是在10μm至30μm范围内其D50粒径呈现出高斯(Gaussian)分布的多元(plural)颗粒。
具体地,可被分级为D50粒径为10μm以上且小于13μm的颗粒、13μm以上且小于14μm的颗粒、14μm以上且小于15μm的颗粒、15μm以上且小于16μm的颗粒及16μm以上且小于30μm的颗粒。
进一步,所述分级后的按D50粒径的组成可以均匀。这从分级所述多元(plural)镍系锂金属氧化物颗粒时,分级后的每个按D50粒径的高斯分布图表与分级前的高斯分布图表具有相同的倾向性得到支持。更为具体的依据请见后述评价。
镍系锂金属氧化物颗粒的内部组成
具体地,所述分级后的按照D50粒径的平均组成可由下述化学式4表示。
[化学式4]Lil+m[Nil-w4-x4-y4-z4Cow4M1x4M2y4M3z4]1-mO2-p4Xp4
在所述化学式4中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w4、x4、y4、z4及p4为满足0<w4≤0.2、0<x4≤0.2、0≤y4≤0.2、0≤z4≤0.1、0<w4+x4+y4+z4≤0.4、0≤p4≤0.1的值,m为满足-0.05≤m≤0.25的值。
这表示按所述分级后的D50粒径,镍的平均摩尔含量为60%以上的高浓度。
例如,在所述化学式4中,M1可以为Mn,0<w4≤0.1,0<x4≤0.1,y4=z4=p4=0,0<w4+x4+y4+z4≤0.2。
具体地,所述镍系锂金属氧化物颗粒内的核部的组成在整个区域可由下述化学式5表示。
[化学式5]Li1+m[Ni1-w5-x5-y5-z5Cow5M1x5M2y5M3z5]1-mO2-p5Xp5
在所述化学式5中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w5、x5、y5、z5及p5为分别满足0≤w5≤0.1、0≤x5≤0.1、0≤y5≤0.1、0≤z5≤0.1、0≤w5+x5+y5+z5≤0.2、0≤p5≤0.1的值,m为满足-0.05≤m≤0.25的值。
这表示在所述镍系锂金属氧化物颗粒内的核部的整个区域中,镍的摩尔含量为80%以上,其水平高于所述平均组成。
例如,在所述化学式5中,M1可以为Mn,0<w5≤0.05,0<x5≤0.05,y5=z5=p5=0,0<w5+x5+y5+z5≤0.1。
而且,所述镍系锂金属氧化物颗粒内的壳部的组成在所述边界面中可由所述化学式5表示,在所述最外廓中可由下述化学式6表示,从所述边界面到所述最外廓,镍(Ni)、M1、M2及M3的各摩尔含量可以逐渐变化。
[化学式6]Li1+m[Ni1-w6-x6-y6-z6Cow6M1x6M2y6M3z6]1-mO2-p6Xp6
在所述化学式6中,M1、M2及M3分别为从包括Mn、Al、Mg、Zr、Sn、Ca、Ge、Ga、B、Ti、Mo、Nb及W的组中选择的一种元素,X为从包括F、N及P的组中选择的一种元素,w6、x6、y6、z6及p6为满足0<w6≤0.3、0<x6≤0.3、0≤y6≤0.1、0≤z6≤0.1、0<w6+x6+y6+z6≤0.5、0≤p6≤0.1的值,m为满足-0.05≤m≤0.25的值。
这表示在所述镍系锂金属氧化物颗粒内与核部的边界面上镍的摩尔含量为80%以上,在所述最外廓上镍的摩尔含量为60%以上,从所述边界面到所述最外廓具有镍的摩尔含量逐渐减少的浓度梯度。与此同时,除所述镍之外的金属(即,M1、M2及M3)从所述边界面到所述最外廓具有镍的摩尔含量逐渐增加的浓度梯度。例如,在所述化学式6中,M1可以为Mn,0<w6≤0.25,0<x6≤0.25,y6=z6=p6=0,0<w6+x6+y6+z6≤0.5。
所述镍系锂金属氧化物颗粒根据所述化学式4的整体组成及所述化学式5的核部组成能够表现高容量的同时,根据所述化学式6的壳部组成能够具有稳定的结构。
涂层
另外,所述正极活性物质可进一步包括涂层,所述涂层围绕所述壳部的外表面并包括B、Mg、Zr、Al、Mn、Co或其组合的元素、所述元素的氧化物、非晶质化合物、锂离子导电性氧化物(例如,硼酸锂(Lithium borate)、硼硅酸锂(lithium borosilicate))、高分子等。
此时,由包含在所述涂层中的物质能够抑制所述镍系锂金属氧化物颗粒与电解质的直接接触及其引起的副反应。
正极活性物质的制备方法
本发明的又一个实施方案提供一种锂二次电池用正极活性物质的制备方法,该方法包括:分别制备第一金属盐水溶液及第二金属盐水溶液的步骤,所述第一金属盐水溶液及第二金属盐水溶液包括镍原料物质、异种金属原料物质及水且所述镍原料物质的摩尔浓度彼此不同;第一共沉淀步骤,以恒定的速度向保持恒定的pH且被供应螯合剂的反应器中供应所述第一金属盐水溶液以形成核部;及第二共沉淀步骤,在所述第一共沉淀步骤之后,逐渐减少所述第一金属盐水溶液的供应速度的同时,逐渐增加所述第二金属盐水溶液供应速度,以形成围绕所述核部的外表面的壳部;所述方法在所述第二共沉淀步骤中获取由所述核部及所述壳部构成的镍系金属氢氧化物颗粒,并控制所述第一共沉淀步骤及所述第二共沉淀步骤,以使下述式2的值成为12.5%以上,在所述第二共沉淀步骤之后,包括烧成所述镍系金属氢氧化物颗粒及锂原料物质的混合物以获取镍系锂金属氧化物颗粒的步骤。
[式2]log(T1)/log(T1+T2)≈R1/(R1+D1)
在所述式2中,T1为第一共沉淀步骤的执行时间,T2为所述第二共沉淀步骤的执行时间。
本发明的一个实施方案中提供的正极活性物质的制备方法包括:混合上述正极活性物质前体和锂原料物质且根据情况添加掺杂原料物质并混合之后烧成的一系列工序。在此,所述正极活性物质前体的制备方法与上述说明相同。
下面,为了除去与上述内容重复的说明,仅说明所述第二共沉淀步骤之后的步骤。
烧成步骤(镍系锂金属氧化物颗粒的获取步骤)
在所述第二共沉淀步骤之后,烧成所述镍系金属氢氧化物颗粒及锂原料物质的混合物,以获取镍系锂金属氧化物颗粒。
此时,如上所述,可向所述混合物添加掺杂原料物质,所述获取的镍系锂金属氧化物颗粒将包括掺杂剂。
所述烧成温度可以为700至800℃,烧成时间可以为12至20小时,这是常见的烧成温度及时间。
涂层形成步骤
在获取所述镍系锂金属氧化物颗粒的步骤之后,可进一步包括形成围绕所述镍系锂金属氧化物颗粒外表面的涂层的步骤。
具体地,形成围绕所述镍系锂金属氧化物颗粒外表面的涂层的步骤可包括:混合所述镍系锂金属氧化物颗粒及涂层原料物质的步骤;及热处理所述镍系锂金属氧化物颗粒及涂层原料物质的混合物的步骤。
所述涂层原料物质可以为B、Mg、Zr、Al、Mn、Co或它们的组合元素、所述元素的氧化物、非晶质化合物、锂离子导电性氧化物(例如,硼酸锂(Lithium borate)、硼硅酸锂(lithiumborosilicate))、高分子等原料物质。混合所述镍系锂金属氧化物颗粒及涂层原料物质的步骤并不局限于干式混合或湿式混合方式。
锂二次电池
本发明的再一个实施方案提供一种锂二次电池,该电池包括:含有上述锂二次电池用正极活性物质的正极;负极;及电解质。
这相当于上述含有正极活性物质从而表现优异性能的锂二次电池,对于所述正极活性物质已进行详细的描述,故省略更为详细的说明。
而且,除所述正极活性物质之外的锂二次电池的结构与常见锂二次电池的结构相同。
下面,描述本发明的较佳实施例及试验例。但是,下述实施例仅是本发明的一个较佳实施例,本发明并不限定于下述实施例。
实施例1(正极活性物质前体)
1)金属盐溶液的制备
首先,作为镍系原料物质利用NiSO4·6H2O,作为钴原料物质利用CoSO4·7H2O,作为锰原料物质利用MnSO4·H2O,制备Ni、Co及Mn的浓度彼此不同的两种金属盐水溶液。
关于用于形成核部的第一金属盐水溶液的制备,在蒸馏水内以满足(Ni0.98Co0.01Mn0.01)(OH)2的化学计量摩尔比的方式混合所述各种原料物质,并使整体金属盐的摩尔浓度成为2.5M。
与此独立地,关于用于形成壳部的第二金属盐水溶液的制备,在蒸馏水内以满足(Ni0.64Co0.23Mn0.13)(OH)2的化学计量摩尔比的方式混合所述各种原料物质,并使整体金属盐的摩尔浓度成为2.5M。
2)共沉淀工序
准备有两个金属盐水溶液供应槽串联而成的共沉淀反应器,并分别向金属盐水溶液供应槽中装入所述第一金属盐水溶液及所述第二金属盐水溶液。
向所述共沉淀反应器(容量为20L,旋转电机的输出功率为200W)装入3升蒸馏水之后,以2升/分钟的速度供应氮气,由此除去溶解氧,将反应器的温度保持50℃的同时,以140rpm的速度进行搅拌。
而且,将作为螯合剂的14M浓度的NH4(OH)以0.06升/小时的速度连续投入反应器中,并将作为pH调节剂的8M浓度的NaOH溶液以0.1升/小时的速度连续投入反应器中,并且适当地控制其投入量,使工序进行中的反应器内保持pH12。
从所述串联的两个金属盐水溶液供应槽向如此保持恒定的pH并被供应螯合剂的反应器中投入金属盐溶液,并且如图1中表示的“核部75%”,调节各金属盐溶液的投入时间及投入量。
具体地,将所述第一金属盐水溶液以0.4升/小时的速度投入的同时,将反应器的叶轮速度调节为140rpm,并将共沉淀反应执行至沉淀物的直径成为约11.1μm。此时,调节流量使溶液在反应器内的平均滞留时间成为10小时左右,反应达到正常状态之后,向所述反应物赋予正常状态持续时间,以获取密度更高的共沉淀化合物。
接着,改变所述第一金属盐水溶液和所述第二金属盐水溶液的混合比率的同时,以整体0.4升/小时的速度投入供应溶液,并将所述第一金属盐水溶液的供应速度逐渐降低为0.05升/小时,将所述第二金属盐水溶液的供应速度逐渐增加为0.35升/小时。此时,调节流量使溶液在反应器内的平均滞留时间成为20小时以内,将共沉淀反应执行至最终沉淀物的直径成为14.8μm。
3)后处理工序
对通过上述一系列共沉淀工序获取的沉淀物进行过滤,并用水清洗之后,在100℃的烤箱(oven)内烘干24小时,获取颗粒整体中的组成为(Ni0.88Co0.095Mn0.025)(OH)2的多元颗粒以作为实施例1的活性物质前体。
实施例2(正极活性物质)
将实施例1中获取的正极活性物质前体及锂盐LiOH·H2O(三田化学电池级(battery grade))用1∶1.05(前体∶锂盐)的摩尔比混合,并将其装入管式炉(tube furnace)(内径50mm,长度1000mm)中,以200mL/min的速度导入氧气的同时进行烧成。
此时,作为烧成条件,在480℃中保持5h,然后在750℃中保持16h,升温速度为5℃/min。而且,作为掺杂材料混合ZrO2粉末,以在所述烧成中使Zr均匀地分布于颗粒内部,并在烧成后干式混合H3BO3,然后进行热处理以使B均匀地涂布于颗粒表面。
最终获取在颗粒整体中的组成为Li1.05(Ni0.88Co0.095Mn0.025)0.996Zr(0.004)O2的颗粒表面上以800ppm涂布有B的多元颗粒,以作为实施例2的活性物质。
比较例1(正极活性物质前体)
1)金属盐溶液的制备
用于形成核部的第一金属盐水溶液及用于形成壳部的第二金属盐水溶液分别使用与实施例1相同的水溶液。
2)共沉淀工序
从所述串联的两个金属盐水溶液供应槽向与实施例1相同的反应器中投入金属盐溶液,并且如图1中表示的“核部<50%”,调节各金属盐溶液的投入时间及投入量。
具体地,将所述第一金属盐水溶液以0.4升/小时的速度投入的同时,将反应器的叶轮速度调节为140rpm,并将共沉淀反应执行至沉淀物的直径成为约6.5μm。此时,调节流量使溶液在反应器内的平均滞留时间成为5小时左右,反应达到正常状态之后,向所述反应物赋予正常状态持续时间,以获取密度更高的共沉淀化合物。
接着,改变所述第一金属盐水溶液和所述第二金属盐水溶液的混合比率的同时,以整体0.4升/小时的速度投入供应溶液,并将所述第一金属盐水溶液的供应速度逐渐降低为0.15升/小时,将所述第二金属盐水溶液的供应速度逐渐增加为0.25升/小时。此时,调节流量使溶液在反应器内的平均滞留时间成为20小时以内,将共沉淀反应执行至最终沉淀物的直径成为13.5μm。
3)后处理工序
对通过上述一系列共沉淀工序获取的沉淀物,与实施例1同样地进行后处理,从而获取颗粒整体中的组成为(Ni0.88Co0.095Mn0.025)(OH)2的多元颗粒,以作为比较例1的活性物质前体。
比较例2(正极活性物质)
对于比较例1的活性物质前体,执行与实施例2同样的工序,从而获取在Li1.05(Ni0.88Co0.095Mn0.025)0.996Zr(0.004)O2的颗粒表面上以800ppm涂布有B的多元颗粒,以作为比较例2的活性物质。
评价例1(正极活性物质粒径分布分析)
将实施例2及比较例2的各1kg的正极活性物质均匀分散于2kg的乙醇(99.9%)中之后,通过按颗粒直径的重量差,分离出含有大量的相对轻的低粒径颗粒的上澄液,按D50粒径进行了分级。
图2a及图2b分别为对实施例2及比较例2的各种正极活性物质进行的粒度分析(PSA,particle size analyze)结果。而且,图3及图4分别为在实施例2及比较例2中分级后的按直径的SEM照片。
从图2a及图3a至图3e可以确认,实施例2的正极活性物质被分级为D50粒径为10μm以上且小于13μm的颗粒(图3a)、13μm以上且小于14μm的颗粒(图3b)、14μm以上且小于15μm的颗粒(图3c)、15μm以上且小于16μm的颗粒(图3d)、以及16μm以上且小于30μm的颗粒(图3e),并且按各个粒径呈现高斯分布。此时,可确认实施例2的按各个粒径呈现的高斯分布具有与未分级颗粒(标为“参考”)相同的倾向性。
与此相反,从图2b及图4a至图4d可以确认,比较例2的正极活性物质被分级为小于10μm的颗粒(图4a)、10μm以上且小于12μm的颗粒(图4b)、12μm以上且小于14μm的颗粒(图4c)、以及14μm以上且小于30μm的颗粒(图4d),并且按各个粒径呈现高斯分布。但是,可确认比较例2的按各个粒径呈现的高斯分布具有与未分级颗粒(标为“参考”)不同的倾向性。
评价例2(按正极活性物质粒径的内部组成分析)
表1及表2记录的是针对实施例2及比较例2的各正极活性物质进行的ICP(电感耦合等离子体)检测结果。
参见表2,在比较例2中,Ni的最大组成差(Cmax-Cmin)为3.3%,表示相对高的值,Co及Mn的组成差分别为1.4~1.0%,也表示较高的值。
而且,在比较例2中掺杂剂Zr在直径小于10um的颗粒中,相比初始设计值过量存在4倍以上,显示出随着颗粒直径增加掺杂量减少的不均匀性。而且,B涂层仅存在于直径小于10um的颗粒中,直径为10um以上的颗粒中并没有检测出B涂层。
与此相反,在实施例2中,可确认Ni的最大组成差为1.71%,其为比较例2的1/2,Co和Mn的组成差也相对较少。这起因于核部的半径被控制为颗粒半径的75%。
而且,在实施例2中可以看出与其颗粒的直径无关地均匀存在Zr和B。但是,Zr和B相较于初始设计值少量减少是因为分级时一部分被溶解于乙醇。
[表1]
[表2]
评价例3(按正极活性物质粒径的电池充放电容量分析)
表3及表4记录的是运用评价例2中分级前及分级后的各种正极活性物质制备电池并对各种情况下的充放电容量进行分析的结果。
为此,针对评价例2中分级前及分级后的各种正极活性材料,按94∶3∶3(活性物质∶粘合剂∶导电材料)的重量比混合粘合剂聚偏二氟乙烯(PVDF,Polyvinylidene Fluoride)和导电材料电化碳黑(商业名:super p)并将其涂布于铝集电体后进行烘干,并进行辊压(roll press)制备电极。
将锂金属(Li-metal)作为对电极使用,将碳酸亚乙酯(EC,Ethylene Carbonate)∶碳酸二甲酯(DMC,Dimethyl Carbonate)的体积比为1∶2的混合溶媒中溶解有1摩尔LiPF6溶液的液体作为电解液使用。
使用所述各组成要素,按照通常的制备方法分别制备2032半电池(halfcoin cell)。
针对各电池,在常温(25℃)中以0.1C的恒电流条件下实施初始充电(4.25V)及放电(2.5V),接着在0.2C下执行充电/放电,此时的充电容量和放电容量记录在表3及表4中。此时,达到充电终止电压时保持其电压,当达到1/20C电流量时转换为放电模式。
在表3及表4中记录在这种条件下进行充放电循环的结果。表3及表4的结果与表1及表2的结果相关。
具体地,从表4可知,由于比较例2的按颗粒直径的Ni含量差异,充电容量和放电容量的偏差较大。特别是,在粒径小于10μm的比较例2中电池放电容量大幅减少,正如表2中所确认,是因为集中包含着掺杂剂Zr。通常,Zr有助于改善热稳定性,但是存在若其掺杂量增加则容量减少的缺点。
与此相反,根据表3,在实施例2中粒径小于13μm时,具有最小210.73mAh/g的放电容量,在粒径超过16μm时,最大放电容量为216.89mAh/g,由此可知偏差较小。
[表3]
[表4]
评价例4(按正极活性物质粒径的电池的寿命特性分析)
表5及表6记录的是运用评价例2中分级前及分级后的各种正极活性物质制备电池并对各种情况下的寿命特性进行分析的结果。
按照与评价例3同样的方式制备各个电池,并在0.3C的恒定的充电速度下对各个电池进行30次充放电循环。在常温(25℃)中进行充放电循环,其结果记录在表5及表6中。
从表6可知,比较例2的按颗粒直径的初始容量偏差严重,在粒径相对小的情况下,30次循环后的容量保持率高,但是直径大于14μm的情况下容量减少较大。
与此相反,从表5可知,实施例2的按颗粒直径的初始放电容量的偏差相对少,并且在30次循环之后也是93%~96%,可见容量保持率非常好。
[表5]
[表6]
评价例5(正极活性物质DSC热分析)
对于实施例2和比较例2中分级前(用Reference标记)及分级后的各种正极活性物质,进行基于差示扫描量热计(differential scanning calorimeter,DSC)的热特性测量,将其结果示于表7、表8、图5a及图5b中。
从图5b及表8可知,比较例2中按颗粒直径的DSC的峰值(peak)温度偏差相对较大。特别是,直径小于10um的颗粒的峰值温度如表8所示大幅降低至195.8℃,但发热峰值(peak)大幅抑制到90.6J/g,这是因为对提高热稳定性作出贡献的Zr的掺杂量远高于具有其他直径的颗粒。而且,从DSC测量结果来看,随着分级后的颗粒大小增加,结晶化开始(on-set)温度和峰值(peak)温度都逐渐增加。
而且,当颗粒大小增加时,如表2所示,Ni的平均摩尔%也会增加,因此根据DSC测量的发热特性应该相对减少,但在表8中结晶化开始(on-set)温度和峰值(peak)温度反而都增加。由此可以推测,当颗粒大小增加时,壳表面部的浓度梯度变得陡峭,从而受到更大的影响。
另一方面,参见图5a及表7,从DSC测量结果来看,实施例2中按颗粒直径的峰值(peak)温度差并不大,几乎均匀。特别是,与比较例2中各种相近的粒径相比较时,可以确认结晶化开始(on-set)温度和峰值(peak)温度都比较高。特别是需要关注发热量,可以看出在实施例2中发热量的数值远远低于比较例2的数值。
因此,从DSC测量结果可以确认,实施例2的热稳定性远高于比较例2的热稳定性。
[表7]
[表8]
评价例6(正极活性物质DS-IR增加率测量)
针对实施例2和比较例2测量在利用分级前的正极活性物质进行充电及放电循环时的直流内阻(Direct Current,Internal resistance:DC-IR),并将其增加率示于图6中。DC-IR为根据循环进展的高速充电可能性的指标,近年来制备电动汽车用电池时,其作为正极材料的规格项目被严格管理。
这种DC-IR特性在高温下测量时能够明确地区分出效果。
因此,在保持45℃恒温的恒温室中进行测量,随着充电及放电循环的进行,假设4.25V充电为满充电(100%)状态,测量了放电电流施加后60秒以后的电压。
之后,将初始DC-IR值换算为0之后进行30次循环,然后将DC-IR值的增加率换算为百分比来标记。
如比较例2,当用于形成核(core)部的共沉淀工序的保持时间较短时,按颗粒大小的镍的浓度差较大。因此,在运用比较例2的正极活性物质的锂二次电池的特性评价中,也能确认DC-IR增加率在第30次中大幅增加为约100%。
相反,在实施例2中,即便将充电及放电循环进行30次之后,DC-IR值的增加率也是约60%左右,可见远远低于比较例2。
由此可知,当运用核部长度增加且具有在壳部中的镍的摩尔含量急剧减少的浓度梯度的正极活性物质时,对锂二次电池的性能表现非常有效。
本发明并不限于上述实施例,可由不同的多种形式制造。本发明所属技术领域的技术人员应能理解,在不改变本发明技术思想或必要特征的基础上可由其他具体形式实施本发明。因此应该理解,以上所述的实施例在所有方面是示意性的,而不是限制性的。
- 还没有人留言评论。精彩留言会获得点赞!