一种含有吡啶并嘧啶类衍生物或其可药用盐的药物组合物的制作方法
本发明属于药物制剂领域,具体涉及一种含有6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮或其药理学上可接受盐的组合物。
背景技术:
乳腺癌是女性最常见的恶性肿瘤之一,具有发病率高,颇具侵袭性,但病程进展缓慢,中国人口协会2010年2月1日在北京发布了《中国乳腺疾病调查报告》,报告显示,我国城市地区乳腺癌的死亡率增长了38.91%,乳腺癌已经成为对妇女健康威胁最大的疾病,目前在研和上市的乳腺癌药物至少有156种,其中68%为靶向治疗药物,大量研究发现肿瘤与细胞周期反常相关,肿瘤细胞中有丝分裂信号蛋白的大量突变和抗有丝分裂信号蛋白缺陷导致增殖紊乱;同时大部分肿瘤都存在基因组不稳定性(gin)和染色体组不稳定性(cin),这三种基本的细胞周期缺陷都直接或间接由cdks的失控引起。周期素依赖性蛋白激酶(cdk,cyclindependentkinase)抑制剂日益成为热门靶标。
目前开发的一代二代cdk抑制剂很多,最受关注的二代药物包括pfizer公司和onyx公司共同开发的cdk4/6抑制剂pd-0332991,其通过抑制cdk4/6的活性,抑制rb的磷酸化,使e2f-rb复合物留滞在胞浆中,阻断细胞周期的启动。临床试验结果(nct00721409)显示,来曲唑单药治疗的患者的无进展存活期(progression-freesurvival,pfs)为7.5月,而来曲唑和pd-0332991药物联用治疗的患者其无进展存活期则延长至26.1月,这一显著优势获得了广泛关注。
wo2014183520公开了与pd-0332991结构相似的一系列满足式(i)通式的cdk4/6抑制剂,具有显著的cdk4/6的抑制活性和高度选择性,并预期这些化合物可能用于一系列肿瘤,并且可以与一系列现有的抗肿瘤剂联合使用,其中包括如下所示的式a化合物,其化学名为6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮:
申请人在先提交的pct/cn2016/070636记载了上述化合物的羟乙基磺酸盐及其i晶型。
然而上述文献均没有公开如何获得稳定,且溶出满足要求的含有上述化合物的药物组合物,研究发现,上述化合物稳定性较差,一些常规处方无法保证组合物的稳定性;同时,在多种常规辅制成的组合物中溶出不佳。因此,需要深入研究,发现其稳定的组合物,并且溶出良好。
技术实现要素:
本发明的目的在于提供一种稳定性良好的药物组合物,并且该药物组合物制备工艺简单,更适合工艺化大生产。另一方面,本发明还提供了一种溶出完全的药物组合物。
本发明提供的药物组合物包含活性药物成分和填充剂,活性药物成分为6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮或其药理学上可接受的盐,所述填充剂含有甘露醇。
本发明中提供的药物组合物中,所述活性成分的药理学上可接受的盐可以优选自活性成分的羟乙基磺酸盐。基于组合物的总重量,所述活性成分的含量范围可以是基于组合物总重量剂计5%-50%;优选10%-35%,更优选为25%-35%。
本发明中提供的药物组合物中,所述填充剂除甘露醇外,还可以含有微晶纤维素、预胶化淀粉、磷酸氢钙中的一种或几种,优选微晶纤维素。在优选的实施方案中,所述填充剂为甘露醇和微晶纤维素的混合物。
本发明的组合物中,填充剂的含量没有特别限制,可以为所述药物组合物的总重的20-90%,优选30%-70%,更优选为40%-65%,最优选为45%-60%。其中甘露醇和微晶纤维素的重量比例为2:1至10:1,优选2.5:1至5:1,最优选2.5:1至3:1。在本发明的一个实施例中,所用甘露醇和微晶纤维素的比例为3:1,可以显著提高样品的稳定性以及溶出速度和程度。
本发明中提供的药物组合物中,可以含有崩解剂,所述崩解剂可以为交联羧甲基纤维素钠、羧甲基淀粉钠、低取代羟丙基纤维素及交联聚维酮中的一种或多种,优选所述崩解剂不含金属元素,更优选交联聚维酮和/或低取代羟丙纤维素。基于组合物的总重量,所述崩解剂含量为约1-30%,优选8%-15%。在本发明的一个实施例中,所用崩解剂为低取代羟丙纤维素,可以显著提高溶出的速度和最终程度。
本发明中提供的药物组合物中,还可含有粘合剂,例如粘合剂为聚乙烯吡咯烷酮、预胶化淀粉、羟丙甲纤维素及羟丙基纤维素中的一种或几种,基于组合物的总重量,所述粘合剂含量为约0.5-10%,优选为0.5%-5%。
本发明中提供的药物组合物中,本发明提供的药物组合物还可包含一种或多种润滑剂,有助于灌装胶囊或压片。基于组合物的总重量,润滑剂可选自滑石粉、硬脂酸镁、硬脂酸锌、山嵛酸甘油酯等。润滑剂的含量为约为0.5%-5%。
在特别优选的实施方案中,本发明提供了一种药物组合物,含有以重量计的如下成分:
1)10%-35%的(6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮或其药理学上可接受的盐;
2)20%-70%的甘露醇;
3)5%-30%微晶纤维素;
4)8%-15%的崩解剂,选自低取代羟丙基纤维素或交联聚维酮中的一种或两种;
5)0.5%-5%的粘合剂,选自预胶化淀粉、羟丙甲纤维素中的一种或两种;
6)任选地0.5%-5%的润滑剂,选自硬脂酸镁、硬脂酸、山嵛酸甘油酯中的一种或几种。
本发明的药物组合物可以采用本领域常见的方法制备,例如高剪切湿法制粒、干法制粒、一步制粒等方法制备药物组合物颗粒,然后压制成片剂。
本发明提供的药物组合物由于采用了含有甘露醇的填充剂,提高了其稳定性,本发明的组合物置于温度40℃、相对湿度75%的环境下,放置1个月、2个月或3个月后,然后采用hplc法测定,有关物质含量不超过1%。
另一方面,本发明的组合物溶出十分完全,根据中国药典2015版二部附录溶出度测定第二法(桨法),以纯化水作为溶出介质,优选1000ml的纯化水,并在37±0.5℃下以50rpm的桨速对本发明组合物进行溶出试验,在45分钟或60分钟溶出度大于等于95%。
附图说明
图1显示实施例1-4的片剂在纯化水中的溶出曲线。
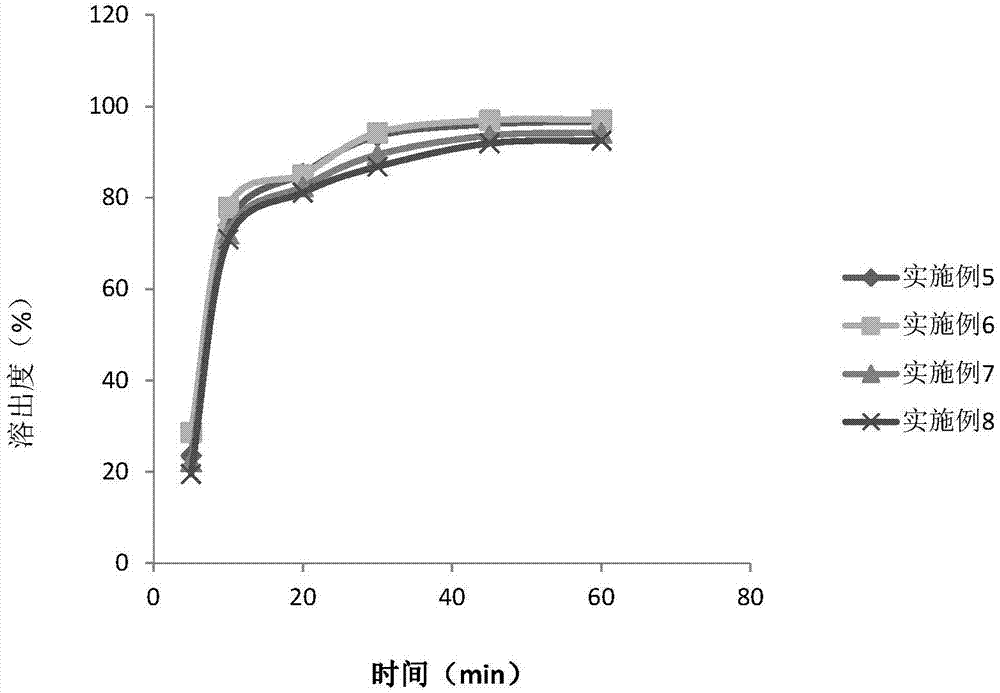
图2显示实施例5-8的片剂在纯化水中的溶出曲线。
图3显示实施例5以及9-12的片剂在纯化水中的溶出曲线。
图4显示实施例6以及13-14的片剂在纯化水中的溶出曲线。
具体实施方式
通过以下实施例和实验例进一步详细说明本发明。这些实施例和实验例仅用于说明性目的,而并不用于限制本发明的范围。
实施例1-4
将6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐(以下简称化合物a)、甘露醇、微晶纤维素、低取代羟丙纤维素、按表1中的比例,采用高速剪切制粒机进行湿法制粒,以羟丙甲纤维素e5配制成的3%的水溶液为润湿剂,对湿软材进行湿整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入处方量的硬脂酸镁,混合均匀。将得到的总混颗粒压制成片剂。
表1
实验例1:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),对实施例1~4的片剂进行溶出度测定。使用1000ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,实施例1~4中,化合物a溶出完全。溶出数据见表2,溶出曲线见图1。
表2
实施例5-8
将6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐(以下简称化合物a)、乳糖或甘露醇、微晶纤维素、低取代羟丙纤维素、按表3中的比例,采用高速剪切制粒机进行湿法制粒,以聚维酮k30配制成的5%的水溶液为润湿剂,对湿软材进行湿整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入处方量的硬脂酸镁,混合均匀。将得到的总混颗粒压制成片剂。
表3
实验例2:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),对实施例5~8的片剂进行溶出度测定。使用1000ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,实施例5~6中,化合物a溶出完全;实施例7~8中,微晶纤维素的比例逐渐增加,与实施例6中的片剂相比较,化合物a的溶出速度逐渐变慢且溶出不完全。溶出数据如下表4所示,溶出曲线见图2。
表4
实施例9-12
将6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐(以下简称化合物a)、乳糖、微晶纤维素、低取代羟丙纤维素或交联聚维酮、交联羧甲基纤维素钠、羧甲基淀粉钠、按表5中的比例,采用高速剪切制粒机进行湿法制粒,以聚维酮k30配制成的5%的水溶液为润湿剂,对湿软材进行湿整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入处方量的硬脂酸镁,混合均匀。将得到的总混颗粒压制成片剂。
表5
实验例3:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),对实施例9~12中的片剂进行溶出度测定。使用1000ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,实施例9~10中,化合物a溶出缓慢且不完全;实施例11中,化合物a完全溶出。实施例12中,化合物a完全溶出,溶出速度较实施例5中片剂慢。溶出数据如下表6所示,溶出曲线见图3。
表6
实施例13~14
将6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐(以下简称化合物a)、甘露醇、微晶纤维素、低取代羟丙纤维素,按表7中的比例,采用高速剪切制粒机进行湿法制粒,分别以聚维酮k30配制成的5%的水溶液和羟丙甲纤维素e5配制成的3%的水溶液、以预胶化淀粉配制成的13%的水溶液作为为润湿剂,对湿软材进行湿整粒及干燥处理,然后将干颗粒(水分小于3%)进行干整粒,加入处方量的硬脂酸镁,混合均匀。将得到的总混颗粒压制成片剂。
表7
实验例4:溶出实验
根据中国药典2015版二部附录溶出度测定第二法(桨法),对实施例6和13、14的片剂进行溶出度测定。使用1000ml的纯化水作为溶出介质,并在37±0.5℃下以50rpm的桨速进行溶出试验。结果表明,实施例6中,化合物a完全溶出;实施例14中,化合物a溶出不完全;实施例13中,化合物a完全溶出。溶出数据如下表8所示,溶出曲线见图4。
表8
实验例5:稳定性研究
将实施例5、6置于温度40℃、相对湿度75%的环境下,放置1个月、2个月、3个月,然后分别采用hplc法测定有关物质的变化。
有关物质检测结果表明,实施例5中片剂的起始有关物质总量大于实施例6中片剂的有关物质总量;实施例5中片剂放置1个月后,有关物质增加;实施6中片剂放置1、2、3个月后,有关物质没有明显变化(见表9)。
表9
实施例15:6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐的合成
步骤1:6-((6-(1-丁氧基乙烯基)-8-环戊基-5-甲基-7-羰基-7,8-二氢吡啶并[2,3-d]嘧啶-2-基)氨基)-5',6'-二氢-[3,4'-联吡啶]-1'(2'h)-甲酸叔丁酯的制备
氩气保护下,将(10g,29.06mmol)2-氨基-6-(1-丁氧乙烯基)-8-环戊基-5-甲基吡啶并[2,3-d]嘧啶-7(8h)-酮(按wo2014183520公开方法制备)、碳酸铯(14.22g,43.75mmol)、pd2(dba)3(2.12g,2.31mmol)、4,5-双二苯基膦-9,9-二甲基氧杂蒽(2.69g,4.69mmol)和125.00g二氧六环投入三口反应瓶中,搅拌均匀后加热至回流,缓慢滴加原料4-(6-氯吡啶-3-基)-5,6-二氢吡啶-1(2h)-羧酸叔丁酯(10.34g,35.00mmol,购自盐城市瑞康医药化工有限公司)和二氧六环(65.62g,0.74mol)的混合液(滴加时间约5h)。滴毕继续回流搅拌反应1~1.5h,tlc监控原料2-氨基-6-(1-丁氧乙烯基)-8-环戊基-5-甲基吡啶并[2,3-d]嘧啶-7(8h)-酮反应完全(展开剂:石油醚:乙酸乙酯=2:1,原料rf=0.6,产物rf=0.7),终止反应。反应液冷却至室温,过滤,滤饼用二氯甲烷17.19g×3洗涤。将滤液于65℃减压浓缩干。残留物加入137.50g二氯甲烷溶解,加入56.25g纯化水,分液,水相再用68.75g二氯甲烷萃取。合并有机相,无水硫酸钠干燥,过滤,滤饼用23.44g二氯甲烷洗,滤液于45℃减压浓缩至油状液体。加入150g丙酮溶解,室温搅拌约2h,冰水浴搅拌约3h。过滤,滤饼用冷丙酮25g×4洗涤,室温减压干燥8~10h,得固体约14.84g,收率:80~92%,hplc检测纯度不低于90%。esi/ms:[m+h]=601.43。
步骤2:4-(6-((6-乙酰基-8-环戊基-5-甲基-7-羰基-7,8-二氢吡啶并[2,3-d]嘧啶-2-基)氨基)吡啶-3-基)哌啶-1-甲酸叔丁酯的制备
将6-((6-(1-丁氧基乙烯基)-8-环戊基-5-甲基-7-羰基-7,8-二氢吡啶并[2,3-d]嘧啶-2-基)氨基)-5',6'-二氢-[3,4'-联吡啶]-1'(2'h)-甲酸叔丁酯(14.84g,24.69mmol)和75g乙酸投入三口反应瓶中,通氩气保护。加入10%pd/c(5g),氢气置换三次,搅拌下于50~60℃常压加氢反应30~32h。hplc法监测中间态剩余量(6-((6-(1-丁氧基乙烯基)-8-环戊基-5-甲基-7-羰基-7,8-二氢吡啶并[2,3-d]嘧啶-2-基)氨基)-5',6'-二氢-[3,4'-联吡啶]-1'(2'h)-甲酸叔丁酯脱掉正丁基保护但双键未被还原的中间体)<0.3%,终止反应。反应液冷却至室温,体系氩气置换后过滤,滤饼用37.50g二氯甲烷洗涤。将滤液于65℃减压浓缩至干。残留物氩气保护用50g无水乙醇回流打浆0.5h,搅拌下自然冷却至室温,冰浴搅拌约4h。过滤,滤饼用冷无水乙醇12.50g×2洗涤。所得湿品加入二氯甲烷31.25g搅拌,过滤不溶物,滤液搅拌下缓慢加入异丙醇118.75g,冰浴搅拌约3h,过滤后减压干燥8~10h得固体约8.75g,产率:60~72%,hplc检测纯度不低于98%。esi/ms:[m+h]=547.26。
步骤3:6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮羟乙基磺酸盐的制备
将4-(6-((6-乙酰基-8-环戊基-5-甲基-7-羰基-7,8-二氢吡啶并[2,3-d]嘧啶-2-基)氨基)吡啶-3-基)哌啶-1-甲酸叔丁酯(8.75g,15.94mmol)和56.25g无水甲醇投入三口反应瓶中,搅拌均匀。将80%羟乙基磺酸(8.81g,55.94mmol)和水0.94g溶于13.75g无水甲醇中,滴加到上述溶液中,溶液变澄清。滴毕加热回流搅拌反应3~3.5h,tlc检测原料反应完全(石油醚:乙酸乙酯=1:1,原料rf=0.3,产物rf=0),终止反应,趁热过滤。滤液搅拌下滴加三乙胺(4.00g,39.38mmol),滴毕继续搅拌约1h,冰浴搅拌约3h。过滤,滤饼用冷无水甲醇7.19g×2洗涤,40℃减压干燥6~8h得固体约7.97g,收率:82~93%,hplc检测纯度不低于98%。
tof-ms:[m+h]=447.2503(6-乙酰基-8-环戊基-5-甲基-2-((5-(哌啶-4-基)吡啶-2-基)氨基)吡啶并[2,3-d]嘧啶-7(8h)-酮结合一个氢离子的离子峰)。
- 还没有人留言评论。精彩留言会获得点赞!