一种载药微球及其制备方法和应用与流程
本发明涉及药物制备领域,具体地,涉及一种载药微球及其制备方法和应用。
背景技术:
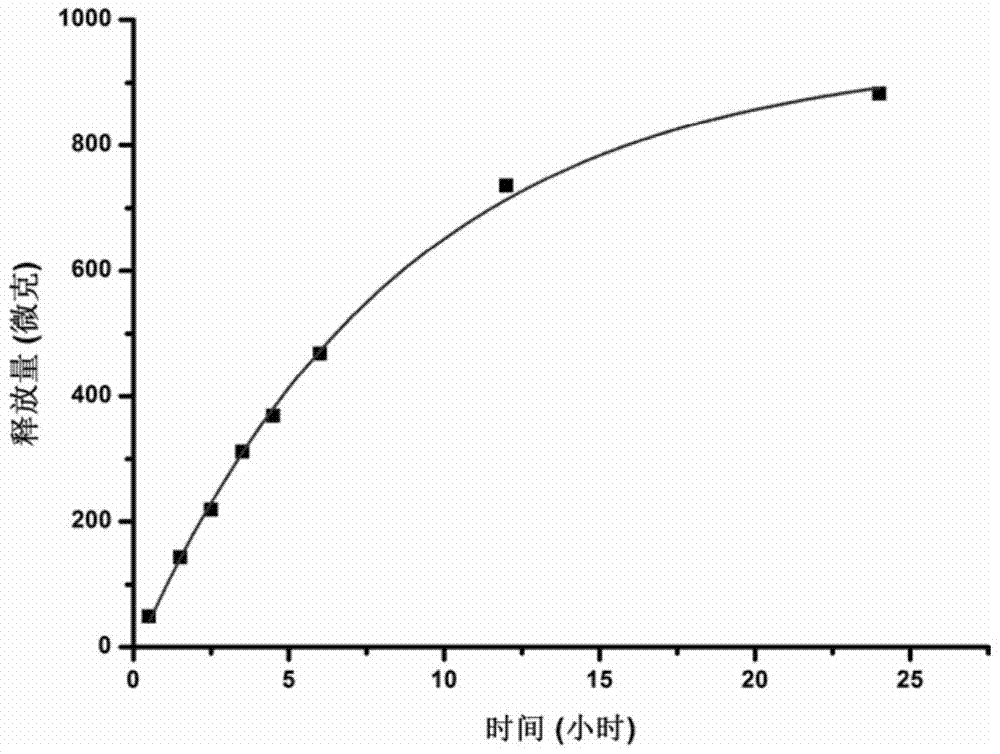
:载药微球一般指粒径尺度为1-500微米的球形颗粒,其主要包括两种组成成分,一是微球的骨架材料,另一是微球包载的原料药。载药微球需要与人体组织接触才能够在临床中发挥药效,因此骨架材料一般是生物相容性良好且可在人体中被降解的材料,包括人工合成的高分子聚合物以及天然高分子聚合物等,上述两类材料在药典中均有详细的描述,作为辅料被广泛的应用在生物医药领域中。载药微球延长了药物的释放周期,并降低了毒副作用,因此可以作为注射剂、口服剂或局部组织植入剂的主要成分,目前主要应用在病菌感染、肿瘤、糖尿病以及抑郁症等疾病的治疗中。由于原料药以及骨架材料的物理化学性质不同,其相应载药微球的制备工艺也有所差异,现行的微球制备工艺主要包括以下几种:第一种为静电喷雾法。静电喷雾又称电纺技术,电纺过程中,原料药与骨架材料溶解分散在有机溶液中,在静电力的作用下形成“taylor”液锥,在不考虑重力的情况下“taylor”液锥受到静电力作用与反方向的表面张力作用。当静电力突破表面张力束缚时,“taylor”液锥逐渐被拉长并在电场下形成微米级的液滴,有机相挥发之后即形成微球。静电喷雾法优点是工序简单,但是走向产业化还有很大的困难。第二种为喷雾干燥法。制备过程中,将聚合物与原料药溶解于水相或者有机相,通过喷雾器使药物溶液迅速变成微米级的液滴,其中形成的液滴与热气体(空气或者氮气)接触使溶剂挥发,从而得到聚集于干燥塔底部的干燥颗粒。喷雾干燥法在食品工业中应用广泛,但应用于载药微球的制备过程中,其产率与药物缓释效果的优化还需要探索。第三种是乳液交联法。乳液交联法一般分为离子交联法与戊二醛交联法,制备过程中,首先将聚合物与原料药溶解于水相,并加入到含有表面活性剂的油相中通过搅拌、超声等形成反相乳液。根据骨架材料的性质不同,可以选择加入阳离子(如钙离子)或者戊二醛溶液进行交联,交联后的微球通过离心、洗涤、干燥即可获取。本方法适用于可以溶于水的骨架材料,但有机相的去除比较困难。第四种为乳液溶剂挥发法。该制备方法分为一步乳化法以及复乳法。一步乳化法以水包油类乳液为主,骨架材料一般是合成的高分子聚合物,原料药溶解或者分散在油相中。复乳法制备过程中,药物水溶液首先在油相中形成初乳,然后初乳再次在水相中形成复乳,本方法适合于包载水中溶解度较高的原料药或者有活性的生物大分子。本方法制备载药微球已经在医药工业中广泛应用,但仍有一些问题需要解决,例如缓释速度控制的工艺以及降低药物突释的工艺等。技术实现要素:本发明的目的是为了克服现有技术中药物缓释速度难以控制的缺陷,提供一种可以有效控制药物缓释速度的载药微球及其制备方法和应用。为了实现上述目的,一方面,本发明提供了一种制备载药微球的方法,该方法包括:(1)将表面活性剂与水混合,制得水相;(2)将药物、药物载体用聚合物材料、第一溶剂和第二溶剂混合,制得油相;(3)将油相加入到水相中进行混合;其中,所述第一溶剂与所述第二溶剂不同,且所述第二溶剂与所述第一溶剂互溶,所述第二溶剂为c1-c3的醇、c1-c3的脂肪酸、c1-c3的脂肪酸酯、c1-c2的醚和乙腈中的至少一种,所述第一溶剂的用量大于所述第二溶剂的用量。第二方面,本发明提供了上述方法制备的载药微球。第三方面,本发明提供了上述载药微球在制备用于治疗肿瘤以及粘膜相关的疾病的药物中的应用。本发明方法制备的载药微球的外观为球形颗粒。采用本方法制备的载药微球,其药物缓释效果不仅可以通过第二溶剂进行控制,0.5小时内的药物突释量也比目前报道的数值要少,药典中“微囊、微球与脂质体制剂指导原则”中规定在开始的0.5小时内释放量要求低于40%,本发明工艺中加入第二溶剂后所制备的微球均达到了要求。本发明的方法制成的微球可以作为栓剂、粘膜冲洗液(如阴道冲洗液)、泡腾片、软膏、粉剂以及药膜的组成成分,并应用于需要持续给药的相关疾病,其中,利用注射、口服、外用或植入等给药方式,将载药微球与人体的粘膜组织、血液或者间质组织结合,从而应用到病菌感染、肿瘤等疾病的治疗中去。上述相关疾病例如与呼吸道粘膜、消化道粘膜、泌尿系统粘膜或生殖系统粘膜相关的肿瘤、上皮组织内病变或炎症。本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。附图说明图1是本发明实施例1制得的载药微球扫描电镜图;图2是本发明实施例1制得的载药微球药物累计释放量曲线图;图3是本发明实施例2-4制得的载药微球药物累计释放量曲线图;图4是本发明实施例5-7制得的载药微球药物累计释放量曲线图;图5是本发明实施例8-10制得的载药微球药物累计释放量曲线图;图6是本发明实施例11-13制得的载药微球药物累计释放量曲线图;图7是本发明实施例14-16制得的载药微球药物累计释放量曲线图;图8是本发明实施例17-19制得的载药微球药物累计释放量曲线图;图9是本发明实施例20-21制得的载药微球药物累计释放量曲线图;图10是本发明实施例22制得的载药微球药物累计释放量曲线图;图11是对比例1制得的载药微球药物累计释放量曲线图。具体实施方式以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。一方面,本发明提供了一种制备载药微球的方法,该方法包括:(1)将表面活性剂与水混合,制得水相;(2)将药物、药物载体用聚合物材料、第一溶剂和第二溶剂混合,制得油相;(3)将油相加入到水相中进行混合;其中,所述第一溶剂与所述第二溶剂不同,且所述第二溶剂与所述第一溶剂互溶,所述第二溶剂为c1-c3的醇、c1-c3的脂肪酸、c1-c3的脂肪酸酯、c1-c2的醚和乙腈中的至少一种,所述第一溶剂的用量大于所述第二溶剂的用量。根据本发明所述的方法,步骤(2)中,优选地,所述第二溶剂为甲醇、乙醇、丙醇、异丙醇、丙二醇、乙酸、乙酸乙酯、乙醚和乙腈中的至少一种,更优选为甲醇、乙酸、乙醇、乙醚、乙腈和异丙醇中的至少一种,从而能够显著提高对药物缓释性的有效控制。根据本发明所述的方法,步骤(2)中,第一溶剂可以为本领域各种有机溶剂,例如可以为二氯甲烷、丙酮、四氢呋喃、氯仿、二氯乙烷、正己烷和二甲基甲酰胺中的至少一种,优选为二氯甲烷、氯仿和四氢呋喃中的至少一种,更优选为二氯甲烷,从而能够与第二溶剂相互配合,进而显著提高对药物缓释性的有效控制。本发明的发明人在研究中发现,第二溶剂可以溶解于水中或挥发性较强。根据第二溶剂粘度的不同,其对药物缓释效果的调控呈现出两个不同的特点:当第二溶剂粘度大于第一溶剂时,随着第二溶剂在油相体系中比例的增加,药物释放的总量也增加,但当第二溶剂在油相体系中比例增加到一定程度时,药物释放的总量反而减小;当第二溶剂粘度小于第一溶剂时,随着第二溶剂在油相体系中比例的增加,药物释放的总量随之降低。前两种情况之下,药物释放的总量整体上均比没有加第二溶剂时的药物释放量要大。根据本发明所述的方法,步骤(1)中,表面活性剂的浓度可以为本领域常规的浓度,例如,水相中表面活性剂与水的质量体积百分数为0.1%-10%,优选为0.5%-5%。即步骤(1)制得的水相是表面活性剂的水溶液,该水溶液配制方法为本领域的常规方法,例如可以包括:将表面活性剂缓慢加入到水中,然后在400-500r/min转速下搅拌10-15min,再将该混合溶液在90-100℃下加热10-15min,然后降温至室温。根据本发明所述的方法,步骤(1)中,表面活性剂的种类可以为本领域各种常规的表面活性剂,例如可以为o/w型乳化剂,优选地,所述表面活性剂为聚乙烯醇和/或聚乙烯吡咯烷酮,从而能够提高药物在制得的载药微球中的分散性。其中,表面活性剂的重均分子量可以为本领域常规的分子量范围,例如可以为20000-30000g/mol。根据本发明所述的方法,步骤(2)中,药物载体用聚合物材料可以为本领域各种药物载体,例如可以为聚己内酯、聚乳酸、聚氨酯、明胶、聚丙烯酸、羧甲基纤维素钠、聚卡波非、壳聚糖、聚乙烯吡咯烷酮、聚乳酸-羟基乙酸共聚物和聚乙烯醇中的至少一种。为了进一步提高对药物缓释性的有效控制,优选地,所述药物载体用聚合物材料为聚乳酸和/或聚乳酸-羟基乙酸共聚物。其中,所述药物载体用聚合物材料为聚乳酸-羟基乙酸共聚物时,其乳酸与羟基乙酸的单体配比可以为75/25或者50/50。根据本发明所述的方法,步骤(2)中,优选地,所述药物载体用聚合物材料的重均分子量为10000g/mol以上,更优选为30000-300000g/mol,从而能够提高药物在药物载体用聚合物材料中的分散性,进一步提高对药物缓释性的有效控制。根据本发明所述的方法,步骤(2)中,所述第一溶剂的用量只要大于所述第二溶剂的用量即可,优选地,所述第一溶剂的体积用量与所述第二溶剂的体积用量比例为1:0.005-0.5,优选为1:0.01-0.1,从而能够进一步提高对药物缓释性的有效控制。根据本发明所述的方法,步骤(2)中,优选地,相对于10ml第一溶剂,所述药物的用量为0.01-2g,所述药物载体用聚合物材料的用量为0.3-3g,更优选地,相对于10ml第一溶剂,所述药物的用量为0.03-1g,所述药物载体用聚合物材料的用量为0.5-2g,从而能够进一步提高对药物缓释性的有效控制。根据本发明所述的方法,步骤(2)中,药物、药物载体用聚合物材料、第一溶剂和第二溶剂混合顺序没有特别地限定,只要混合后形成均匀的油相溶液或者悬浮液即可。例如,当药物与第一溶剂互容时,可以将药物、药物载体用聚合物材料、第一溶剂和第二溶剂一起混合,当药物与第一溶剂不互容时,可以先将药物与第一溶剂混合,再将得到的混合液与药物载体用聚合物材料混合,最后再加入第二溶剂。在一种具体的实施方式中,将药物与第一溶剂在400-600r/min转速下混合10-15h,然后加入药物载体用聚合物材料溶解,再加入第二溶剂混合均匀。根据本发明所述的方法,步骤(3)中,更优选地,步骤(1)得到的水相和步骤(2)得到的油相的体积比为1:0.01-0.15。根据本发明所述的方法,步骤(3)中,所述混合的条件可以为本领域常规的混合条件,例如可以包括:温度为-5~25℃,搅拌速度为200-2000转/分钟;优选地,温度为-5~5℃,搅拌速度为400-1000转/分钟;从而更有利于形成载药微球,进而进一步提高对药物缓释性的有效控制,其中搅拌的时间可以根据实际需要调整,只要能够使得水相和油相混合形成微球,且第一溶剂和第二溶剂挥发完全即可。根据本发明所述的方法,在一种优选实施方式中,步骤(3)中,在100-200r/min搅拌速度下,将油相加入到水相中,然后在400-1000r/min搅拌速度下搅拌1-2h,最后将混合液固液分离,得到载药微球。其中,固液分离的方式可以为离心分离或者过滤分离。根据本发明所述的方法,该方法还可以包括:将得到的载药微球进行干燥处理,其中,所述干燥处理的方式可以为本领域的常规方式,例如可以根据药物载体用聚合物材料与药物的物理化学性质选择以下方式进行干燥。一是在真空干燥箱或者鼓风干燥箱中烘干,温度一般设置为25℃到45℃。二是利用冷冻干燥机进行冻干,冻干之前,将微球放置于-80℃的冰箱中进行预冷,使微球冻住,然后利用已经预冷的冷冻干燥机在真空条件下进行冻干。本发明中,所述药物可以为本领域各种常规的药物,例如可以为抗肿瘤药、消炎药、止痛药、抗生素、抗过敏药以及抗真菌药中的至少一种,更优选地,所述药物为5-氟尿嘧啶、阿司匹林、布洛芬、甲硝唑、西替利嗪和硝酸咪康唑中的至少一种,最优选为5-氟尿嘧啶。根据本发明所述的方法,其中,所述药物的大小可以为本领域各种常规的尺寸,例如药物的平均粒径可以小于50μm,优选为0.05-20μm,从而能够提高药物在药物载体用聚合物材料上的分散性,进而进一步提高对药物缓释性的有效控制。第二方面,本发明提供了上述方法制备的载药微球。本发明制得的载药微球的平均粒径可以为60-400μm,载药微球的药物包封率可以高达55-90%。本发明中,载药微球表面形貌定性以及定量检测的方法如下:将制备的载药微球粘附于导电胶上,并将导电胶固定于电子显微镜样品台上,在高倍下观察微球的尺寸以及微球表面的形貌信息,利用电子显微镜自带软件测量微球的尺寸。载药微球的药物包载率测试方法和原理如下:将载药微球溶解于可溶解高分子支架材料的溶剂中,然后加入与所述溶剂不互溶的药物溶剂中进行药物的萃取,取萃取液利用酶标仪、紫外分光光度计或液相色谱仪测定药物浓度,药物包载率等于单位质量微球中实际的药物含量与理论含量之比。例如溶解高分子支架材料的溶剂可以为二氯甲烷,与二氯甲烷不互溶的药物溶剂可以为水。载药微球的药物释放效率测试方法可以如下:将载药微球分散于ph7.4的磷酸盐缓冲液中,在37℃摇床条件下进行药物释放,每隔一段时间取一定体积的释放液,然后补加相同体积的ph7.4的磷酸盐缓冲液,连续测定一段时间。计算药物的累计释放量并绘制药物的释放曲线。第三方面,本发明提供了上述的载药微球在制备用于治疗肿瘤以及粘膜相关的疾病的药物中的应用。根据本发明所述的应用,可以采用各种常规的方法将上述载药微球作为栓剂、粘膜冲洗液(如阴道冲洗液)、泡腾片、软膏、粉剂以及药膜的组成成分,并应用于需要持续给药的相关疾病,其中,利用注射、口服、外用或植入等给药方式,将载药微球与人体的粘膜组织、血液或者间质组织结合,从而应用到病菌感染、肿瘤等疾病的治疗中去,上述相关疾病例如与呼吸道粘膜、消化道粘膜、泌尿系统粘膜或生殖系统粘膜相关的肿瘤、上皮组织内病变或炎症。实施例1本实施例用于说明本发明的载药微球及其制备方法和应用。(1)水相制备:称取2g聚乙烯醇pva(重均分子量为20000g/mol,购自acrosorganics),缓慢加入100ml水中,然后在500r/min转速下搅拌10分钟,再将聚乙烯醇与水的混合液在90℃水浴锅中,水浴加热10分钟后取出,放置1h将温度降至室温备用。(2)油相制备:将90mg5-氟尿嘧啶(5-fu)与3ml二氯甲烷在500r/min转速下搅拌混合12h,然后再加入450mg左旋聚乳酸plla(重均分子量为80000g/mol,购自济南岱罡生物工程有限公司)涡旋溶解1分钟,再加入150μl无水乙醇,涡旋混匀1min。(3)微球制备:将3ml上述制得的油相倒入烧杯100ml水相中,利用冰浴将液体温度控制在0℃,使用磁力搅拌器以200r/min的转速进行低速搅拌,边低速搅拌边将制得的油相加入水相中。随后将搅拌速度提高至800r/min,持续搅拌1h。然后收集混合液,利用离心的方法进行固液分离,收集微球颗粒,将该微球颗粒在30℃鼓风干燥箱中烘干,制备成载有5-fu的左旋聚乳酸微球。(4)载有5-fu的左旋聚乳酸微球用于制备治疗肿瘤以及粘膜相关的疾病的药物。对比例1按照实施例1的方法制备载药微球,不同的是,不加入第二溶剂无水乙醇。实施例2本实施例用于说明本发明的载药微球及其制备方法和应用。(1)水相制备:称取2g聚乙烯醇(重均分子量为30000g/mol,购自acrosorganics),缓慢加入100ml水中,然后在500r/min转速下搅拌10分钟,再将聚乙烯醇与水的混合液在90℃水浴锅中,水浴加热10分钟后取出,放置1h将温度降至室温备用。(2)油相制备:将60mg5-氟尿嘧啶(5-fu)与3ml二氯甲烷在500r/min转速下搅拌混合12h,然后再加入450mg聚乳酸-羟基乙酸共聚物plga(重均分子量为40000g/mol,单体比例为75/25,购自济南岱罡生物工程有限公司)涡旋溶解1分钟,再加入30μl异丙醇,涡旋混匀1min。(3)微球制备:将3ml上述制得的油相倒入烧杯100ml水相中,利用冰浴将液体温度控制在3℃,使用磁力搅拌器以200r/min的转速进行低速搅拌,边低速搅拌边将制得的油相加入水相中。随后将搅拌速度提高至600r/min,持续搅拌2h。然后收集混合液,利用离心的方法进行固液分离,收集微球颗粒,将该微球颗粒在30℃鼓风干燥箱中烘干,制备成载有5-fu的聚乳酸-羟基乙酸共聚物微球。(4)载有5-fu的聚乳酸-羟基乙酸共聚物微球用于制备治疗肿瘤以及粘膜相关的疾病的药物。实施例3-4本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例2的方法制备载药微球,不同的是,异丙醇的用量分别为150μl、300μl,制得载有5-fu的聚乳酸-羟基乙酸共聚物微球。对比例2按照实施例2的方法制备载药微球,不同的是,不加入第二溶剂异丙醇。实施例5-7本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,5-fu的使用量是60mg,将乙醇替换为乙酸,乙酸的用量分别为30μl、150μl,300μl,制得载有5-fu的左旋聚乳酸微球。实施例8-10本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,将乙醇替换为甲醇,甲醇的用量分别为30μl、150μl,300μl,制得载有5-fu的左旋聚乳酸微球。实施例11-13本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,5-fu的使用量是60mg,将乙醇替换为乙腈,乙腈的用量分别为30μl、150μl,300μl,制得载有5-fu的左旋聚乳酸微球。实施例14-16本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,5-fu的使用量是60mg,将乙醇替换为乙醚,乙醚的用量分别为30μl、150μl,300μl,制得载有5-fu的左旋聚乳酸微球。实施例17-19本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,5-氟尿嘧啶(5-fu)的用量为45mg,无水乙醇的用量分别为150μl、240μl,300μl,制得载有5-fu的左旋聚乳酸微球。实施例20-21本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,5-氟尿嘧啶(5-fu)的用量为16mg,二氯甲烷的量为2ml,左旋聚乳酸plla用量为160mg,聚乙烯醇pva用量为1g,无水乙醇的用量分别为20μl、60μl,制得载有5-fu的左旋聚乳酸微球。对比例3按照实施例20的方法制备载药微球,不同的是,不加入第二溶剂无水乙醇。实施例22-23本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,二氯甲烷体积为8ml,5-氟尿嘧啶(5-fu)的用量为128mg,左旋聚乳酸plla用量为1600mg,聚乙烯醇pva用量为4g,无水乙醇的用量分别为80μl、400μl,制得载有5-fu的左旋聚乳酸微球。对比例4按照实施例22的方法制备载药微球,不同的是,不加入第二溶剂无水乙醇。实施例24本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例1的方法制备载药微球,不同的是,第二次的搅拌速度为400r/min,持续搅拌2h。实施例25本实施例用于说明本发明的载药微球及其制备方法和应用。按照实施例2的方法制备载药微球,不同的是,plga分子量为300000g/mol,制备载有5-fu的聚乳酸-羟基乙酸共聚物微球。测试例1.1微球粒径统计将制备的载药微球粘附于导电胶上,并将导电胶固定于电子显微镜样品台上,在高倍下观察微球的尺寸以及微球表面的形貌信息(参见图1),利用电子显微镜自带软件测量微球的尺寸,结果见下表1。1.2微球的包封率测试称取微球质量30mg,加入二氯甲烷适量,搅拌溶解后,加入30ml纯水,以800r/min转速搅拌,蒸发二氯甲烷,直至烧杯中不见液相分层。取部分液体,经0.45um滤膜过滤,使用紫外分光光度计测量266nm处吸光度。根据5-fu吸光度曲线计算溶液中5-fu的浓度n。计算得包封率和微球载药量,结果见下表2。1.3微球的缓释性能测试量取微球11mg置于15ml离心管,加入pbs10ml,旋紧管盖,在37℃水浴摇床中,于放置后不同时间点取出离心管离心,取样,补液。样品在266nm处测得吸光度,根据5-fu吸光度曲线计算药物释放量结果见下表3-11和图1-11。表1实施例载药微球平均粒径(μm)实施例1182μm对比例1185μm实施例2-483.3μm对比例278μm实施例5-7170.9μm实施例8-10225.75μm实施例11-13168.9μm实施例14-16183μm实施例17-19145.7μm实施例20-21130.3μm对比例3125μm实施例22233.7μm实施例23381μm实施例24240μm实施例25173μm对比例4190μm表2表3表4表5表6表7表8表9表10表11通过表3和图2、11可以看出,实施例1制得的载药微球在半小时内药物释放量为3.9%,而对比例1制得的载药微球在半小时内药物释放量为3.75%,比较可以看出,当加入第二溶剂时,能够显著提高对药物缓释效果的控制。通过表4和图3可以看出,实施例2-4制得的载药微球在半小时内药物释放量为2.74%,6.53%,3.83%,并且实施例2-4制得的载药微球在24小时内药物释放的总量随着第二溶剂乙酸体积比的增加而出现相应的变化,且均优于对比例2。通过表5和图4可以看出,实施例5-7制得的载药微球在半小时内药物释放量为1.33%,7.69%,10.02%,并且实施例5-7制得的载药微球在24小时内药物释放的总量随着第二溶剂乙酸体积比的增加而出现相应的变化,且均优于对比例1。通过表6和图5可以看出,实施例8-10制得的载药微球在半小时内药物释放量为3.78%,12.54%,8.83%,并且实施例8-10制得的载药微球24小时内药物释放的总量随着第二溶剂甲醇体积比的增加而出现相应的变化,且均优于对比例1。通过表7和图6可以看出,实施例11-13制得的载药微球在半小时内药物释放量为4.25%,5.06%,5.62%,实施例11-13制得的载药微球24小时内药物释放的总量随着第二溶剂乙腈体积比的增加而出现相应的变化,且均优于对比例1。通过表8和图7可以看出,实施例14-16制得的载药微球在半小时内药物释放量为1.26%,3.80%,8.92%,实施例14-16制得的载药微球24小时内药物释放的总量随着第二溶剂乙腈体积比的增加而出现相应的变化,且均优于对比例1。通过表9和图8可以看出,实施例17-19制得的载药微球在半小时内药物释放量为14.83%,10.40%,15.81%,实施例17-19制得的载药微球24小时内药物释放的总量随着第二溶剂乙醇体积比的增加而出现相应的变化,且均优于对比例1。通过表10和图9可以看出,实施例20-21制得的载药微球在半小时内药物释放量为7.15%,3.28%,24小时内药物释放的总量随着第二溶剂乙醇体积比的增加而出现相应的变化,且均优于对比例3。通过表11和图10可以看出,实施例22和23制得的载药微球在半小时内药物释放量分别为5.89%,8.24%,24小时内药物释放的总量随着第二溶剂乙醇体积比的增加而出现相应的变化,且均优于对比例4。通过表11的数据还可以看出,实施例24制得的载药微球在半小时内药物释放量为4.99%,实施例25制得的载药微球在半小时内药物释放量为0.44%。由图1可以看出,本发明方法制备的载药微球的外观为球形颗粒。采用本方法制备的载药微球,其药物缓释效果不仅可以通过第二溶剂进行控制,0.5小时内的药物突释量也比目前报道的数值要少,药典中“微囊、微球与脂质体制剂指导原则”中规定在开始的0.5小时内释放量要求低于40%,本发明工艺中加入第二溶剂后所制备的微球均达到了要求。本发明的方法制成的微球可以作为栓剂、粘膜冲洗液(如阴道冲洗液)、泡腾片、软膏、粉剂以及药膜的组成成分,并应用于需要持续给药的相关疾病,其中,利用注射、口服、外用或植入等给药方式,将载药微球与人体的粘膜组织、血液或者间质组织结合,从而应用到病菌感染、肿瘤等疾病的治疗中去。上述相关疾病例如可以与呼吸道粘膜、消化道粘膜、泌尿系统粘膜或生殖系统粘膜相关的肿瘤、上皮组织内病变或炎症。以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1