华盖散颗粒的制备方法和质量控制方法与流程
本发明属于药物制备工艺与质量控制领域,涉及华盖散颗粒的制备方法,还涉及华盖散颗粒质量控制方法。
背景技术:
华盖散出自《太平惠民和剂局方》,华盖散具有宣肺解表,祛痰止咳的功用;主治肺感寒邪,咳嗽上气,胸膈烦满,项背拘急,声重鼻塞,头昏目眩,痰气不利,呀呷有声。本方集肺经之药于一身,诸药相伍,使表寒解、肺气宣,痰涎化,喘咳平,肺系诸症皆除,故称“华盖散”。方中以麻黄为君,辛温解表,宣肺平喘以祛风寒。臣以苏子,辛香降气化痰,杏仁辛苦温,化痰止咳平喘,二药共同降利肺气,祛痰止咳。佐药陈皮辛温,燥湿化痰,理气行滞,“气顺则痰消”;桑白皮甘寒,泻肺利水平喘;茯苓健脾渗湿,杜绝生痰之源;三药共同祛湿消痰。甘草为使调和诸药。诸药配伍,解表与化痰并用,痰湿得消,表寒得散,诸证自愈。
经典名方标准颗粒仿照传统中药汤剂的煎煮模式,既保留传统汤剂有效成分的特点,又避免了中成药难以随症加减和传统汤剂煎煮、携带不便的问题。该品种的开发,可形成“以经方标准颗粒为主,单味配方颗粒为辅,随症加减”的临床调剂模式,满足现代临床用药需求。本品的开发可以解决单味配方颗粒存在的无法共煎等争议的问题,充分发挥传统方剂配伍的优势,达到减毒增效的效果,尤为重要。复方配方颗粒不仅传承了中药单味配方颗粒的优点,同时又考虑了饮片在煎煮过程中的相互作用,符合中医传统用药理论,具有较大的价值。
由于我国尚无适合于临床调剂的复方颗粒,目前国际市场销售的中药复方颗粒均来自日本、韩国及我国台湾地区。该品种的研究将推进中药颗粒剂的国际市场开拓,促进中药国际化。因此,有必要在研发并国产华盖散,而研发对华盖散的制备必须明确质量控制方法。有研究文献报道对同处方但不同制法的华盖散传统汤剂进行其中盐酸麻黄碱、苦杏仁苷、甘草酸、甘草苷的含量测定(靳凤云,贺祝英,赵杨,廖薇,梁光义.hplc测定华盖散传统汤剂与颗粒汤剂中盐酸麻黄碱、苦杏仁苷、甘草酸、甘草苷的含量,中成药,2008年第30卷第1期),另有研究报道测定其中的橙皮苷成分(张明昶,邵进明,彭小冰,袁双秀,梁光义.反相高效液相色谱法测定不同制法华盖散中橙皮苷的含量.时珍国医国药,2010年第21卷第1期)。目前报道的上述定量或定性的方法还不能有效地控制华盖散颗粒的质量。
技术实现要素:
有鉴于此,本发明的目的在于提供一种华盖散颗粒的制备方法,该方法能充分发挥中药配伍的特色及传统汤剂的优势,最大限度的保留了有效成分;本发明的目的之二在于提供华盖散颗粒的质量控制方法,该方法在于克服现有质量标准监督的不足之处,针对华盖散颗粒提供了一种系统的检测手段,通过对华盖散颗粒中多指标成分盐酸麻黄碱、盐酸伪麻黄碱、甘草苷、橙皮苷、迷迭香酸的含量测定方法来达到对华盖散颗粒的质量控制。
为达到上述目的,本发明提供如下技术方案:
1、华盖散颗粒的制备方法,包括如下步骤:
a、按质量比为1-3:1-3:1-3:1-3:1-3:1-3:1-3:1-3分别取麻黄、炒苦杏仁、炒紫苏子、陈皮、炙桑白皮、茯苓和炙甘草,然后加入相当于药材总质量6~12倍的水进行第一次煎煮,过滤收集滤液,药渣再加入相当于药材总质量4~10倍的水进行第二次煎煮,过滤收集滤液,药渣再加入相当于药材总质量3-8倍的水进行第三次煎煮,过滤收集滤液;
b、合并3次煎煮滤液,滤过,滤液真空减压浓缩至相对密度为1.12~1.18的浓缩液;
c、以糊精和乳糖的混合物作为底物,喷入步骤b所得浓缩液中,沸腾干燥质粒,筛选粒度在16-60目的颗粒,得华盖散颗粒。
优选的,所述麻黄、炒苦杏仁、炒紫苏子、陈皮、炙桑白皮、茯苓和炙甘草的质量比为3:3:3:3:3:3:1.5。
优选的,所述第一次煎煮时间为1~3小时,所述第二次煎煮时间为1~1.5小时,所述第三次煎煮时间为0.5~1.5小时。
本发明步骤c中,所述糊精和乳糖的质量比为3:1.
本发明中,所述沸腾干燥条件为进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa。
本发明中,所述糊精和乳糖的混合物加入量为干膏量的1.12倍。
2、华盖散颗粒的质量控制方法,包括如下步骤:
(1)盐酸麻黄碱、盐酸伪麻黄碱含量的测定:取华盖散颗粒,研磨后加甲醇溶液,超声溶解,冷却,再加甲醇溶液补足失重,摇匀,过滤,收集滤液得华盖散颗粒样品;盐酸麻黄碱、盐酸伪麻黄碱标准品用甲醇溶液溶解,摇匀得标准品溶液,然后将华盖散颗粒样品和标准品溶液在如下色谱条件下检测:色谱柱:长度和直径为250mm×4.6mm,5μm的kromasil100-5phenyl;柱温:27℃;进样量:10μl;流速:1.0ml·min-1;流动相:乙腈与磷酸质量分数为0.1%的磷酸水体积比为1∶99;检测波长:210nm;
(2)甘草苷、橙皮苷、迷迭香酸含量的测定:取华盖散颗粒研磨后用甲醇溶液,制成华盖散颗粒样品,然后将甘草苷、橙皮苷、迷迭香酸标准品加甲醇溶解,制成标品溶液,再将华盖散颗粒样品和标准品溶液在如下色谱条件下检测:色谱柱:长度和直径250×4.6mm,5μm的thermosyncronisc18;柱温:27℃;流动相:乙腈-0.01%磷酸水;进样量:20μl;流速:1.0ml·min-1;流动相:乙腈与磷酸质量分数为0.1%的磷酸水梯度洗脱,0~12min乙腈体积比为18%,12~30min乙腈体积比为20%,30~40min乙腈体积比为25%,40~55min乙腈体积比为100%;
(3)结果判断:华盖散颗粒样品在盐酸麻黄碱、盐酸伪麻黄碱、甘草苷、橙皮苷和迷迭香酸标品出峰处出峰符合质量标准。
本发明步骤(1)中,华盖散颗粒样品制备方法为:取华盖散颗粒,研磨后按3每克华盖散颗粒加250ml含盐酸的体积分数为50%的甲醇溶液,超声溶解,冷却,再加含盐酸的体积分数为50%的甲醇溶液补足失重,摇匀,过滤,收集滤液得华盖散颗粒样品;所述甲醇溶液中盐酸质量分数为0.036%;
标准品溶液制备方法为:分别称取盐酸麻黄碱、盐酸伪麻黄碱,用体积分数为50%的甲醇溶解并稀释,配制成含盐酸麻黄碱、盐酸伪麻黄碱分别为20μg/ml、10μg/ml的溶液即可。
本发明步骤(2)中,华盖散颗粒样品制备方法为:取华盖散颗粒,按每克华盖散颗粒加100ml甲醇,在频率40khz、功率240w条件下超声提取30min,取出,冷却,补足失重,在10000r·min-1离心10min,上清液用0.45μm微孔滤膜滤过即可;
标准品溶液制备方法为:分别称取甘草苷、橙皮苷、迷迭香酸,加甲醇溶解配制成浓度分别为0.3733mg·ml-1、0.0902mg·ml-1和0.1911mg·ml-1的溶液。干膏量为中药经水煎煮,浓缩后干燥得到的膏体。
本发明的有益效果在于:华盖散颗粒的制备方法,该方法按照传统方法煎煮制成复方颗粒,较目前单味配方颗粒服用时各药味简单相加,更能充分发挥中药配伍的优势,体现了中医药整体的观念,为临床用药提供新的选择;制备工艺中采用沸腾制粒技术,将提取浸膏与辅料混合、制粒、干燥、整粒等多工序,在一台制粒机中完成,改进传统制粒多机联用的工艺弊端,使物料在低温条件下完成干燥、制粒过程,避免有效成分的破坏,保证了药物的疗效。
本发明还公开了华盖散颗粒的质量控制方法,通过多指标含量测定,可更为全面地对华盖散颗粒的质量进行评价,保证产品质量的稳定性及临床用药的有效性与安全性。本发明具有检测手段简单、检测结果准确、质量监督更加全面的特点,更能适合整体中药质量控制提高的要求。
附图说明
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图进行说明:
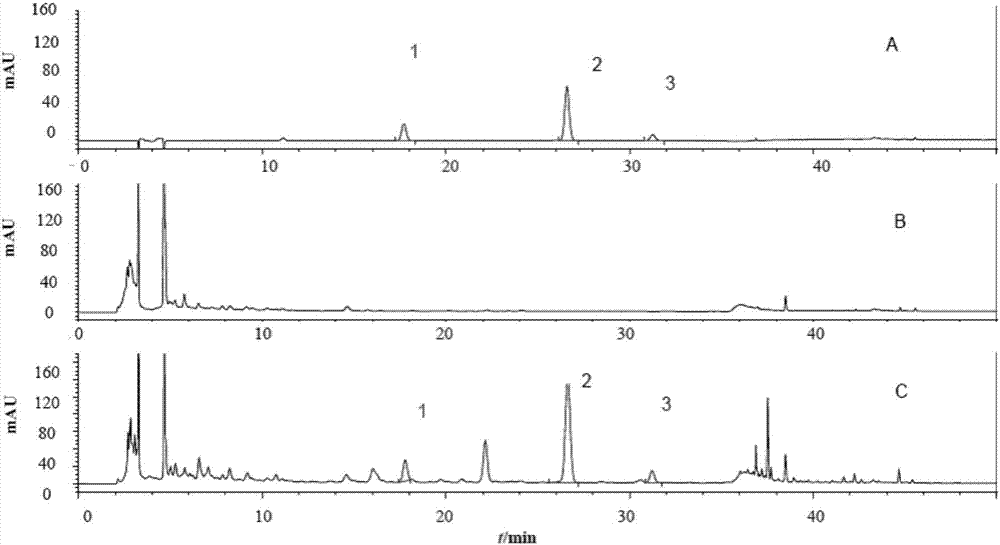
图1为盐酸麻黄碱、盐酸伪麻黄碱专属性考察图谱(a:溶剂;b:盐酸麻黄碱;c:盐酸伪麻黄碱)。
图2为甘草苷、橙皮苷、迷迭香酸专属性考察hplc图谱(a:甘草苷;b:橙皮苷;c:迷迭香酸)。
具体实施方式
下面将结合附图,对本发明的优选实施例进行详细的描述。
实施例1
华盖散颗粒的制备方法,包括如下步骤:
a、取麻黄300g、炒苦杏仁300g、炒紫苏子300g、陈皮300g、炙桑白皮300g、茯苓300g、炙甘草150g,加19.5kg的水,煎煮提取2小时,即一煎;一煎后药渣加入15.6kg的水,煎煮提取1小时,即二煎;二煎后药渣加入11.7kg的水,煎煮提取1.0小时,即三煎;
b、合并三次煎煮滤液,滤过,滤液真空减压浓缩至相对密度为1.14~1.15(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混匀(混合物加入量为干膏量的1.12倍),作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,制得粒度在16-60目的华盖散颗粒。
实施例2
华盖散颗粒的制备方法,包括如下步骤:
a、取麻黄300g、炒苦杏仁300g、炒紫苏子300g、陈皮300g、炙桑白皮300g、茯苓300g、炙甘草150g,加23.4kg的水,煎煮提取2.5小时,即一煎;一煎后药渣加入19.5kg的水,煎煮提取1.5小时,即二煎;二煎后药渣加入15.6kg的水,煎煮提取1小时,即三煎;
b、合并三次煎煮滤液,滤过,滤液真空减压浓缩至相对密度为1.15~1.16(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,制得粒度在16-60目的华盖散颗粒。
实施例3
华盖散颗粒的制备方法,包括如下步骤:
a、取麻黄300g、炒苦杏仁300g、炒紫苏子300g、陈皮300g、炙桑白皮300g、茯苓300g、炙甘草150g,加15.6kg的水,煎煮提取3小时,即一煎;一煎后药渣加入11.7kg的水,煎煮提取1小时,即二煎;二煎后药渣加入7.8kg的水,煎煮提取0.5小时,即三煎;
b、合并三次煎煮滤液,滤过,滤液真空减压浓缩至相对密度为1.17~1.18(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,制得粒度在16-60目的华盖散颗粒。
实施例4
华盖散颗粒的制备方法,包括步骤如下:
a、取麻黄300g、炒苦杏仁300g、炒紫苏子300g、陈皮300g、炙桑白皮300g、茯苓300g、炙甘草150g,加11.7kg的水,煎煮提取1小时,即一煎;一煎后药渣加入7.8kg的水,煎煮提取1小时,即二煎;二煎后药渣加入5.85kg的水,煎煮提取1.5小时,即三煎;
b、合并三次煎煮滤液,滤过,滤液真空减压浓缩至相对密度为1.12~1.13(60℃)的浓缩液;
c、加入重量比为3:1的糊精及乳糖混匀,作为底物,喷入上述浓缩液,沸腾干燥制粒,控制进风温度:80℃、风机频率35hz、进料速度40r·min-1、雾化压力0.1mpa,制得粒度在16-60目的华盖散颗粒。
实施例5、华盖散颗粒含量测定
将实施例1~4制备的华盖散颗粒进行含量测定,采用高效液相色谱法,具体步骤如下:
1、盐酸麻黄碱、盐酸伪麻黄碱的含量测定方法为:
(1)样品、仪器与试剂:
华盖散颗粒(批号:15120001、15120002、15120003、16010001、16010002、16010003、16020001、16020002、16020003、16030001)盐酸麻黄碱对照品(中国食品药品检定研究院,批号110749-200714,质量分数99.8%),盐酸伪麻黄碱对照品(中国食品药品检定研究院,批号110715-200212,质量分数99.9%)。
agilent1100四元梯度泵(美国安捷伦公司)和自动进样器;sartorius-bs124s型精密电子天平(德国赛多利斯天平有限公司);kq2200e型超声清洗机器(上海五相仪器仪表有限公司);超声波清洗仪(kq3200de,昆山市超声仪器有限公司)。
甲醇、乙睛(美国fisher)、三乙胺、二正丁胺、磷酸(天津科密欧化学试剂有限公司);水为重蒸馏水;其余试剂均为分析纯。
(2)方法与结果:
检测波长的选择:精密称取盐酸麻黄碱、盐酸伪麻黄碱对照品10mg、5mg,加甲醇配制盐酸麻黄碱、盐酸伪麻黄碱20μg/ml、10μg/ml。以甲醇为空白校正,于200~400nm范围内扫描,确定盐酸麻黄碱、盐酸伪麻黄碱的最大吸收波长λmax为210nm。
色谱条件的确定:
色谱柱:长度和直径为250mm×4.6mm,5μm的kromasil100-5phenyl;柱温:27℃;进样量:10μl;流速:1.0ml·min-1;流动相:乙腈-0.1(wt)%磷酸水(含0.04(wt)%三乙胺和0.02(wt)%二正丁胺)(1∶99)。
对照品溶液的制备:
精密称取盐酸麻黄碱、盐酸伪麻黄碱对照品适量,加50%甲醇溶解并稀释,将其配制成每ml中分别含盐酸麻黄碱、盐酸伪麻黄碱20μg、10μg混合溶液,摇匀,即得。
供试品溶液的制备:
取华盖散标准颗粒,研细,取细粉约0.3g,置25ml量瓶中,加50%甲醇溶液(0.036%盐酸),超声(500w,40khz)处理30min,取出放冷,加50%甲醇溶液(0.036%盐酸)至刻度,摇匀,离心,取上清液滤过,取续滤液,即得。
(3)专属性研究
取华盖散标准颗粒样品和缺麻黄阴性样品,制成供试品溶液、缺麻黄的供试品溶液。分别吸取三种溶液,照拟定的色谱条件,注入液相色谱仪,记录色谱图,见图1(1-盐酸麻黄碱峰、2-盐酸伪麻黄碱峰;a-缺麻黄的华盖散阴性样品,b-盐酸麻黄碱、盐酸伪麻黄碱对照品,c-华盖散标准颗粒)。
(4)线性关系的考察
分别精密吸取对照品储备溶液置容量瓶中加甲醇定容,形成5个浓度的混合对照品溶液,注入液相色谱仪,记录色谱图,以峰面积为纵坐标,对照品的进样量(μg)为横坐标,按最小二乘法原理拟合方程,绘制标准曲线,盐酸麻黄碱方程为y=2.157x-2.481,r=0.9999;盐酸伪麻黄碱方程为y=2.230x+2.485,r=1.0000。结果表明,盐酸麻黄碱进样量在31~993μg范围内,与相应峰面积线性关系良好;盐酸伪麻黄碱进样量在29~933μg范围内,与相应峰面积线性关系良好。
(5)回收率试验
取本品颗粒(盐酸麻黄碱、盐酸伪麻黄碱含量分别为:1.61mg·g-1、0.60mg·g-1)适量,研细,精密称取9份,分别置25ml量瓶中,再精密加入混合对照品(含盐酸麻黄碱0.02643mg·ml-1,盐酸伪麻黄碱0.01002mg·ml-1)适量,再加50%甲醇溶液(含0.036%盐酸)适量,按供试品制备方法制备供试品溶液,依照拟定的色谱条件,分别吸取10μl供试品溶液、对照品溶液于液相色谱仪,记录色谱图,计算加样回收率,结果如表1和表2。结果表明:本方法检测华盖散标准颗粒中盐酸麻黄碱、盐酸伪麻黄碱含量回收率良好,符合分析要求。
计算公式如下(下同):
表1、盐酸麻黄碱加样回收率试验
表2、盐酸伪麻黄碱加样回收率试验
(6)精密度试验
a.重复性考察
取华盖散标准颗粒适量,研细,称取6份,按供试品溶液制备方法制备,测定峰面积,计算含量,考察结果表明盐酸麻黄碱、盐酸伪麻黄碱rsd分别为0.96%、0.90%,重复性良好。
b.不同人员精密度考察
取华盖散标准颗粒,由3个不同的工作人员,按供试品溶液制备方法分别制备供试品溶液2份,测定其峰面积,计算出含量,结果显示:人员变动的精密度rsd为分别为0.64%、1.64%,表明不同人员精密度良好。
c.不同日期精密度考察
取华盖散标准颗粒,在不同的工作日内,按供试品溶液制备方法分别制备2份,测定峰面积,计算含量,结果不同日期精密度考察,两者日间精密度rsd为1.89%、3.28%,表明日间精密度良好。
d.不同仪器精密度考察
取华盖散标准颗粒,在不同的仪器上,按供试品溶液制备方法分别制备2份,测定峰面积,计算含量,结果不同仪器精密度考察,仪器变动的精密度rsd分别为0.66%、1.64%,表明精密度良好。
(7)稳定性的考察
取华盖散标准颗粒,按供试品溶液制备方法制备供试品溶液,于不同的时间间隔,分别精密吸取10μl,按上述色谱条件,注入液相色谱仪,记录峰面积。结果表明:供试品溶液室温下密闭保存,存放24h,对盐酸麻黄碱、盐酸伪麻黄碱保留时间及峰面积rsd分别为1.49、1.86%,稳定性良好。
(8)盐酸麻黄碱、盐酸伪麻黄碱含量限度的确定
按上述供试品配制方法配制各批次样品供试液,以上法测定10批次华盖散颗粒盐酸麻黄碱、盐酸伪麻黄碱含量。得平均含量分别为:1.43mg/g、0.54mg/g;确定盐酸麻黄碱、盐酸伪麻黄碱的含量限度分别为:1.14mg/g、0.43mg/g,结果见表3。
表3、不同批次华盖散颗粒中盐酸麻黄碱、盐酸伪麻黄碱含量的测定结果
2、甘草苷、橙皮苷、迷迭香酸含量的测定。
(1)样品、仪器与试剂
样品:华盖散颗粒(样品同盐酸麻黄碱、盐酸伪麻黄碱含量测定方法用样品);甘草苷对照品(中国食品药品检定研究院,批号111610-201106,质量分数93.7%),橙皮苷对照品(中国食品药品检定研究院,批号110796-200310,质量分数95.3%),迷迭香酸对照品(中国食品药品检定研究院,批号111871-201404,质量分数98.6%)。
仪器:agilent1100四元梯度泵(美国安捷伦公司)和自动进样器;sartorius-bs124s型精密电子天平(德国赛多利斯天平有限公司);kq2200e型超声清洗机器(上海五相仪器仪表有限公司);超声波清洗仪(kq3200de,昆山市超声仪器有限公司)。
试剂:甲醇、乙睛(美国fisher)、磷酸(天津科密欧化学试剂有限公司);水为重蒸馏水;其余试剂均为分析纯。
(2)方法与结果
色谱条件的确定:色谱柱:thermosyncronisc18(250×4.6mm,5μm);柱温:27℃;流动相:乙腈-0.01%磷酸水;进样量:20μl;流速:1.0ml·min-1;流动相:a(乙腈)-b(0.01(wt)%磷酸水);梯度洗脱程序:0min→12min→30min→40min→55min,乙腈18%→20%→25%→100%→100%。
对照品溶液的制备:精密称取对照品甘草苷、橙皮苷、迷迭香酸适量,分别置25ml、100ml、25ml量瓶中,加甲醇溶解并定容,摇匀,即得对照品储备溶液ⅰ、ⅱ、ⅲ(浓度分别为0.3733mg·ml-1、0.0902mg·ml-1、0.1911mg·ml-1)。
供试品溶液的制备:取适量供试品研细,精密称取约0.25g于具塞三角锥形瓶中,加入25ml甲醇。称重,超声(频率40khz,功率240w)提取30min,取出,放冷,补重,10000r·min-1离心10min,续滤液用微孔滤膜(0.45μm)滤过,作为供试液。
检测波长的选择:精密称取甘草苷、橙皮苷、迷迭香酸对照品适量,加甲醇配制甘草苷、橙皮苷、迷迭香酸(浓度分别为0.3733mg·ml-1、0.0902mg·ml-1、0.1911mg·ml-1)。于200~400nm范围内扫描,确定甘草苷、橙皮苷、迷迭香酸的最大吸收波长λmax为278nm。
(3)专属性研究
取华盖散标准颗粒样品和同时缺甘草、陈皮、紫苏子的阴性样品,按拟定样品处理方法处理,制备成华盖散标准颗粒供试品溶液、同时缺甘草、陈皮、紫苏子的阴性供试品溶液。取三种溶液按拟定色谱条件,分别吸取20μl,注入液相色谱仪,记录色谱图,见图2(1-甘草苷峰,2-橙皮苷峰,3-迷迭香酸峰;a-混合对照品溶液hplc图,b-缺甘草、陈皮、紫苏子药材的阴性样品,c-华盖散标准颗粒样品)。结果表明,同时缺甘草、陈皮、紫苏子的阴性样品溶液在主峰处无干扰,方法专属性良好。
(4)线性范围考察
分别精密吸取对照品储备溶液置容量瓶中加甲醇定容,形成5个浓度的混合对照品溶液,注入液相色谱仪,记录色谱图,以进样量(μg)为横坐标,峰面积为纵坐标,绘制标准曲线,按最小二乘法原理计算甘草苷的回归方程:y=2375.02x-18.545,相关系数r为0.99993(线性范围0.012~0.598μg)、橙皮苷的回归方程:y=2032.02x-60.016,相关系数r为0.99987(线性范围0.043~1.442μg);迷迭香酸的回归方程:y=1928.39x-15.077,相关系数r为0.99997,(线性范围0.012~0.458μg)。结果表明,三者线性关系良好。
(5)回收率试验
取甘草苷、橙皮苷、迷迭香酸混合对照品溶液(浓度分别为:0.0747mg·ml-1、0.6045mg·ml-1、0.0573mg·ml-1)加入已知含量的颗粒中(已测甘草苷、橙皮苷、迷迭香酸含量分别为:0.90mg·ml-1、6.60mg·ml-1、0.64mg·ml-1),按供试品制备方法制备供试品溶液,照供试品溶液制备方法制备,按拟定色谱条件,分别吸取20μl供试品溶液与对照品溶液,注入液相色谱仪,记录色谱图,并计算加样回收率,结果如表4~6。结果表明:本方法检测华盖散标准颗粒中甘草苷、橙皮苷、迷迭香酸含量回收率良好,符合分析要求。
表4、甘草苷加样回收率试验
表5、橙皮苷加样回收率试验
表6、迷迭香酸加样回收率试验
(6)精密度试验
a.重复性考察:取适量样品研细,称取6份,按供试品制备方法制备供试品溶液,分别测定其峰面积,计算出含量,结果rsd为0.62%、0.766%、0.80%,重复性良好。
b.不同人员精密度考察
取华盖散标准颗粒样品,由3个不同的工作人员,按供试品溶液制备方法制备供试品溶液2份,分别测定其峰面积,计算含量,结果显示:人员变动的精密度rsd均小于2%,精密度良好。
c.不同日期精密度考察
取华盖散标准颗粒样品,在不同的工作日内,按供试品溶液制备方法制备供试品溶液2份,分别测定其峰面积,计算含量,结果显示:日间精密度rsd均小于2%,表明日间精密度良好。
d.不同仪器精密度考察
取华盖散标准颗粒,在不同的仪器上,按供试品溶液制备方法分别制备2份,测定峰面积,计算甘草苷、橙皮苷、迷迭香酸含量,结果仪器变动的精密度rsd均小于2%,表明精密度良好。
(7)稳定性试验
取华盖散标准颗粒,按供试品溶液制备方法制备供试品溶液,分别于不同的时间间隔,精密吸取20μl,按上述色谱条件,注入液相色谱仪,记录甘草苷、橙皮苷、迷迭香酸峰面积。结果表明:供试品溶液室温下密闭保存,存放24h,对甘草苷、橙皮苷、迷迭香酸保留时间及峰面积变异均小于2.0%,稳定性良好。
(8)甘草苷、橙皮苷、迷迭香酸含量限度的确定:
精密吸取甘草苷、橙皮苷、迷迭香酸对照品(浓度分别为0.3733mg·ml-1、0.0902mg·ml-1、0.1911mg·ml-1)及供试品溶液20μl,于上述条件下,注入液相色谱仪,测定。
按上述供试品配制方法配制各批次样品供试液,以上法测定10批次华盖散颗粒甘草苷、橙皮苷、迷迭香酸含量。得平均含量分别为甘草苷0.71mg/g、橙皮苷6.71mg/g、迷迭香酸0.31mg/g;确定甘草苷、橙皮苷、迷迭香酸的含量限度为:甘草苷0.57mg/g、橙皮苷5.37mg/g、迷迭香酸0.25mg/g。结果见表7。
表7、不同批次华盖散颗粒中甘草苷、橙皮苷、迷迭香酸含量的测定结果
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!