可用于检测四环素的纳米纤维及其制备和应用的制作方法
本发明属分子检测技术领域涉,具体涉及一种纳米尺度下的可用于检测四环素的分子印迹纳米纤维及其制备方法及应用。
背景技术:
四环素类抗生素具有一定毒性,长期食用含抗生素的食品对肝、肾功能具有一定的损伤,甚至有致癌、致畸、致突变作用,易引起体内耐药菌增加,使抗菌药物失效,因而,其残留检测技术一直是食品安全研究领域的一个重要热点。
目前检测四环素的主要方法有微生物法、薄层色谱法、免疫分析法、毛细管电泳法、高效液相色谱法等,各种方法均有一定的优缺点,欧盟法律对各种动物源性食物中毒四环素(母体及异构体总量)最大残留限量作出如下规定:肌肉中不超过100ppb,肝脏中不超过300ppb,肾脏中不超过600ppb,蛋中不超过200ppb,牛奶中不超过100ppb。我国农业部也规定动物源性食品中四环素类抗生素的最大残留限量为100ppb。
分子印迹技术(MIT)是指把某一个特定的目标分子作为模板分子,制备对该目标分子具有特异选择性聚合物的过程,通常被定义为制备与识别“分子钥匙”的“人工锁”的技术。分子印迹聚合物由于具有与天然抗体同样的识别性能力和与高分子同样的抗腐蚀性能的双重优点,它具有结构预定性、特异识别性和广泛实用性三大特点,因此,它在许多领域,如色谱中对映体和异构体的分离、固相萃取、化学仿生传感器、模拟酶催化、临床药物分析、膜分离技术等领域均展现了良好的应用前景。
对于一些生物样品或环境样品,被测组分浓度通常非常低,因此往往需要对样品进行预处理,进行富集,纯化。固相萃取技术因其具有易于实现自动化、灵活多变以及对环境友好等优点,目前已被视为分析测定前对样品进行预处理的标准方法。为了使被测组分获得高的富集倍数并尽量纯化基本成分,要求用于固相萃取的吸附剂材料对被测物质要有更高的亲和力和选择性。然而传统的吸附剂如C8和C18键合硅胶材料,选择性差,往往难于满足复杂样品的要求;而具有高度选择性的生物活性材料如免疫亲和剂又有制备麻烦、费时、成本高、稳定性差及不宜在有机溶剂中使用等缺点。利用分子印迹化合物的分子识别能力,可望实现有选择的固相萃取,克服生物样品体系复杂、预处理手续繁琐等因素,在富集待分析物的同时,除去大部分干扰组分,从而方便仪器分析。
目前,已有关于采用分子印迹技术分离四环素的相关研究报道,但大多为通过本体聚合制备印迹颗粒,以颗粒的形式直接装填固相萃取柱,尚未有印迹纳米纤维制备固相萃取填充材料的报道。
上述内容改为:目前,已有关于基于分子印迹理论分离四环素的研究,如CN101397163,CN1011080等,其制备的分离核心材料为以四环素为模板,利用本体聚合技术获得的固体颗粒,由于难以控制粒径,导致颗粒的粒径分布范围比较大,粒径不均一,在装填时会不均匀,进而影响分离效果。
技术实现要素:
本发明的目的是在现有技术的基础上,提供一种具有高亲和性和高选择性吸附的分子印迹纤维材料。
本发明的另一目的是提供一种上述分子印迹纤维材料的制备方法。
本发明的第三目的是提供一种纳米纤维在食品中四环素的分离或富集方面的应用。
本发明的目的可以通过以下措施达到:
一种可用于检测四环素的纳米纤维,它由如下步骤制备而成:
(A)将四环素、甲基丙烯酸、二甲基丙烯酸乙二醇酯和乙腈充分混合后,加入引发剂充分混合,在惰性气体保护下密封进行聚合反应;
(B)聚合反应后分离出聚合物,用溶剂洗脱除去四环素,干燥,得到四环素分子印迹微球;
(C)将四环素分子印迹微球与聚苯乙烯、N,N-二甲基甲酰胺和四氢呋喃混合得到纺丝液,采用静电纺丝装置进行静电纺丝,纺出的纤维干燥后得到分子印迹的纳米纤维。
一种可用于检测四环素的纳米纤维的制备方法,它包括如下步骤:
(A)将四环素、甲基丙烯酸、二甲基丙烯酸乙二醇酯和乙腈充分混合后,加入引发剂充分混合,在惰性气体保护下密封进行聚合反应;
(B)聚合反应后分离出聚合物,用溶剂洗脱除去四环素,干燥,得到四环素分子印迹微球;
(C)将四环素分子印迹微球与聚苯乙烯、N,N-二甲基甲酰胺和四氢呋喃混合得到纺丝液,采用静电纺丝装置进行静电纺丝,纺出的纤维干燥后得到分子印迹的纳米纤维。
在上述各步骤(A)中,所述四环素与甲基丙烯酸、二甲基丙烯酸乙二醇酯和引发剂的摩尔比优选为1:4~8:20~40:5~10;所述引发剂为偶氮二异丁氰。
在步骤(A)中,四环素、甲基丙烯酸、二甲基丙烯酸乙二醇酯和乙腈混合后可以进行超声处理或其他能够充分混合的混合方式进行处理。加入引发剂后可继续进行超声处理或其他能够充分混合的混合方式进行处理。研究表明,若四环素与甲基丙烯酸的比例过低,则不利于模板分子与功能单体发生有效聚合,过高则会使得模板分子会泄露,而二甲基丙烯酸乙二醇酯的浓度过低,无法交联,而浓度过高则会导致过度交联,对后续洗脱操作造成困难。
在聚合反应前,可以将已加入引发剂的液体置于容器中,通入氮气置换气体,再进行聚合反应。
在一种优选方案中,聚合反应的温度为50~70℃,反应时间为8~24小时。
一种具体的步骤(A)为:将四环素(模板)、甲基丙烯酸(功能单体)、二甲基丙烯酸乙二醇酯(交联剂),按模板:功能单体:交联剂=1:4~8:20~40的比例(摩尔比)投入到乙腈(致孔剂)中,混合溶解后,加入偶氮二异丁氰(引发剂),混合溶解后,置于密封试管中,通入氮气,在50~70℃热聚合反应8-24小时。
在上述各步骤(B)中,聚合反应后固液分离出固体聚合物,例如可采用离心的方式分离出聚合物。
对聚合物的洗脱可采用索氏提取器进行。洗脱溶剂可以为冰醋酸和甲醇混合溶剂,洗脱至洗脱液中检测不出四环素。在一种优选方案中,洗脱溶剂中甲醇和冰醋酸的体积比为8~10:1。
一种具体的步骤(B)为:将反应得到的聚合物溶液离心沉淀,所得固体装入索氏提取器,用甲醇:冰醋酸=8~10:1(体积比)作为提取溶剂进行提取,直到洗脱液检测不出四环素为止。将提取后的聚合物用甲醇洗涤并干燥,得到四环素分子印迹微球。
除不加模板分子四环素外,基于同样的(A)和(B)的步骤,可得到空白微球。
在上述各步骤(C)中,将分子印迹微球与聚苯乙烯、N,N-二甲基甲酰胺、四氢呋喃混合,加入到注射器中,使用微量注射泵,以一定的流速,通过高压直流静电场,在距离注射器针头一定距离的位置设置铝箔接收器,开始静电纺丝,纺好的丝经干燥后,最终得到分子印迹的纳米纤维。
步骤(C)中四环素分子印迹微球与聚苯乙烯、N,N-二甲基甲酰胺和四氢呋喃的优选质量比为1:5~10:50~100:50~100。
在步骤(C)中,所述纺丝液可以加入到注射器中,使用微量注射器,使纺丝液从注射器针头喷出通过高压直流静电场至所述注射器针头前方的接收器,进行静电纺丝
在步骤(C)的一种优选方案中,静电纺丝时电压为10~18KV,微量注射器的推进速度为0.5~0.8mL/h,接收器与注射器针头的距离在15~20cm。纺丝液中聚苯乙烯的浓度在10%~50%之间。研究表明,聚苯乙烯的浓度低于10%时,导致出丝困难,而浓度大于50%,会产生明显串珠,过高或过低的浓度都会导致纺丝失败,按照本法所纺的纤维直径在300-500纳米之间。
一种具体的步骤(C)的方法为:将分子印迹微球与聚苯乙烯、N,N-二甲基甲酰胺、四氢呋喃以1:5~10:50~100:50~100(质量比)混合,搅拌过夜,将悬浮液加入到注射器中,排掉空气后,将注射器装入到推进器上,同时设置铝箔接收装置,将高压静电纺丝机的电压设置在10~18KV,微量推进器的速度为0.5~0.8mL/h,接收器的距离在15~20cm。开始静电纺丝,直至结束。将获得的纤维置于50℃真空干燥箱内烘干。
用步骤(C)同样的方法采用空白微球可制备空白纤维,用同样的方法不使用分子印迹微球可制备不含微球的纤维。
本发明的纳米纤维在制备后,可将其填充到萃取柱的下端,压实。用甲醇淋洗,依次用乙腈、冰醋酸、双蒸水对固相萃取柱活化处理,处理后的固相萃取柱即可使用。
本发明的纳米纤维可应用在食品中,特别是牛奶、蜂蜜、水产、肉类等食品中,四环素的分离或富集方面的应用。例如本发明的分子印迹纤维材料可用作固相萃取填料,结合高效液相色谱技术实现对动物源食品中兽药残留的分离、富集与测定。
本发明以四环素分子为模板,通过沉淀聚合制备分子印迹微球,通过静电纺丝制备分子印迹纤维,然后将四环素分子印迹纤维填充到固相萃取柱中,得到四环素分子印迹固相萃取柱,再应用四环素分子印迹固相萃取柱,纯化、富集蜂蜜中的四环素残留,与高效液相色谱连用,能够建立一种基于印迹纤维技术的全新的兽用抗生素四环素的检测方法。
本发明的有益效果:
本发明基于分子印迹理论,以纤维为基材,将颗粒负载到纤维上,既具有特异性的识别能力,又能克服颗粒粒径布不均一的缺点,因其具有更大的比表面积,故吸附容量更优越,且制备纤维时用到更少的印迹分子,故成本更低,纤维本身制备工艺成熟,故而有更好的应用前景。
附图说明
图1是本发明所用的四环素高效液相图;
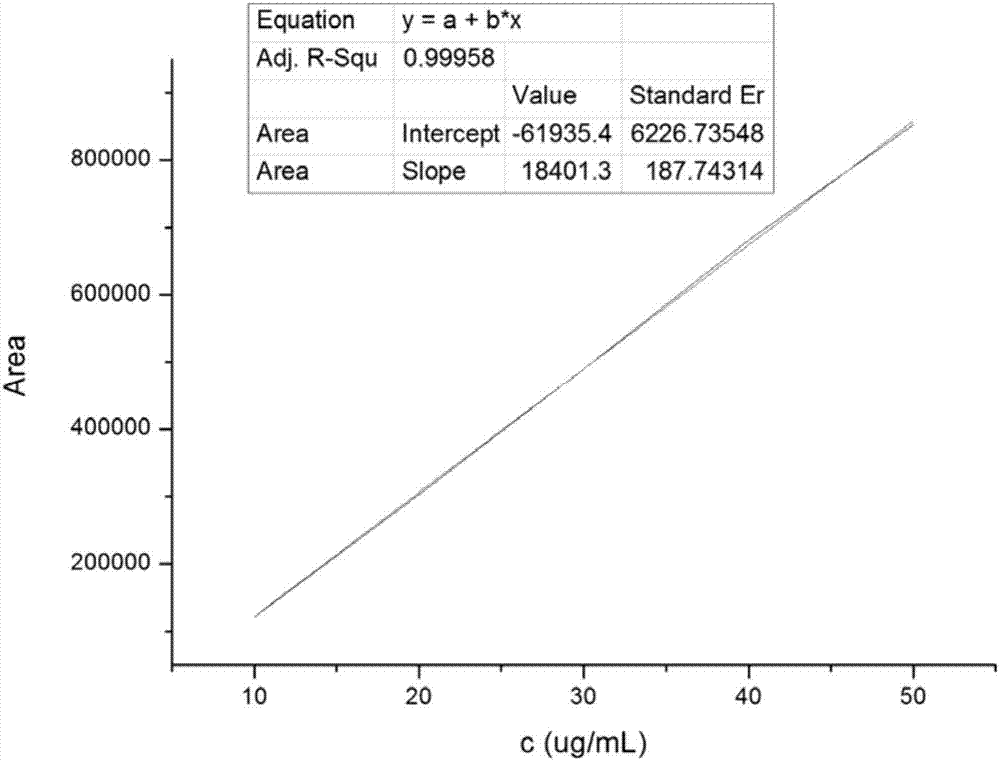
图2是本发明所用的四环素标准曲线图;
图3是本发明实施例1所得的四环素分子印迹微球电镜图;
图4是本发明实施例3所得的四环素分子印迹纤维电镜图。
具体实施方式
下面对本发明的具体实施方式和操作过程做详细说明,本发明的保护范围包含但不限于下述的实施例。
实施例1
将0.0444克的四环素,85微升的甲基丙烯酸,二甲基丙烯酸乙二醇酯940微升,与10毫升乙腈混合,超声10分钟,加入偶氮二异丁氰10毫克,超声10分钟,将溶液倒入试管,通氮气10分钟,密封。置于50℃恒温水浴中反应8小时,得到乳白色浑浊液,离心得到沉淀,甲醇洗涤,50℃真空干燥得到白色微球。将微球装入索氏提取器,用10%的冰醋酸甲醇溶液洗脱,直至洗脱液无四环素为止。用纯甲醇再次洗脱,50℃真空干燥得到脱去模板分子的印迹微球。
实施例2
将140微升的甲基丙烯酸,二甲基丙烯酸乙二醇酯1400微升,与10毫升乙腈混合,超声10分钟,加入偶氮二异丁氰15毫克,超声10分钟,将溶液倒入试管,通氮气10分钟,密封。置于50℃恒温水浴中反应16小时,得到乳白色浑浊液,离心得到沉淀,甲醇洗涤,50℃真空干燥得到空白微球。
实施例3
将分子印迹微球0.1克与聚苯乙烯1克、N,N-二甲基甲酰胺5毫升、四氢呋喃7毫升混合,室温搅拌24小时,将悬浮液加入到针筒注射器中,排掉空气后,将注射器装入到螺旋推进器上,同时距针头15厘米处设置铝箔接收装置,将高压静电纺丝机的正电压设置在18KV,微量螺旋推进器的速度为0.8毫升/小时,控制湿度在50%以下,开始静电纺丝,直至结束。将获得的纤维置于50℃真空干燥箱内烘干。
实施例4
将空白微球0.1克与聚苯乙烯0.5克、N,N-二甲基甲酰胺6毫升、四氢呋喃6毫升混合,室温搅拌24小时,将悬浮液加入到针筒注射器中,排掉空气后,将注射器装入到螺旋推进器上,同时距针头15厘米处设置铝箔接收装置,将高压静电纺丝机的正电压设置在15KV,微量螺旋推进器的速度为0.6毫升/小时,控制湿度在50%以下,开始静电纺丝,直至结束。将获得的纤维置于50℃真空干燥箱内烘干。
实施例5
将聚苯乙烯1克、N,N-二甲基甲酰胺7.5毫升、四氢呋喃7.5毫升混合,室温搅拌24小时,将悬浮液加入到针筒注射器中,排掉空气后,将注射器装入到螺旋推进器上,同时距针头15厘米处设置铝箔接收装置,将高压静电纺丝机的正电压设置在17KV,微量螺旋推进器的速度为0.7毫升/小时,控制湿度在50%以下,开始静电纺丝,直至结束。将获得的纤维置于50℃真空干燥箱内烘干。
实施例6
取自制的固相萃取小柱,将制得的纤维5毫克填充到柱的下端,压实。用1毫升甲醇淋洗,再依次用1ml乙腈、冰醋酸、双蒸水对固相萃取柱活化处理,处理后的固相萃取柱即可使用。
将市售蜂蜜1克,加入5毫升Na2EDTA-Mcllvaine缓冲溶液,涡旋,离心,取上清液,上固相萃取柱,用5毫升5%的甲醇水溶液洗脱,弃去洗脱液,抽干,用乙酸乙酯(0.1毫升*3)洗脱,将滤液过0.22微米滤膜,进高效液相检测。检测仪器:安捷伦1200高效液相色谱,色谱条件:C18ODS柱,检测波长374nm,流动相比例:乙腈:水:磷酸=20:80:0.1,采用外标法,做四环素标准曲线,浓度范围从10μg-50μg/mL。计算加入量分别为1μg和3μg印迹球纤维,空白球纤维、空白纤维、印迹微球和空白微球加标的回收率和RSD(n=3)
表一 实际样品中四环素的测定
- 还没有人留言评论。精彩留言会获得点赞!