环丙烯氨基酸以及方法与流程
本发明涉及经由(扩展的)遗传密码来位点特异性地引入生物正交基团。详细而言,本发明涉及经由遗传引入的氨基酸如赖氨酸来将氨基甲酸酯键合的环丙烯引入多肽。此类环丙烯基对另外的化学基团如四嗪的添加是有用的。
背景技术:
经由遗传密码扩展来位点特异性地引入生物正交基团提供了对用任何探针来位点特异性标记蛋白质的强有力通用策略。然而,可经遗传编码的生物正交官能团的缓慢反应性和/或它们对光激活的需求限制了此策略的效用。
对化学生物学家而言,使用多样的探针来快速、位点特异性标记蛋白质仍是未完成的挑战;酶介导的标记方法可能是快速的,但其使用在研究中的蛋白质内引入扰动的蛋白质或肽融合,并且可能限制可标记的位点;而许多可经遗传编码的组分的‘生物正交’反应太缓慢以致不能在对研究许多生物过程有用的时间标度上实现蛋白质的定量和位点特异性标记。
对在多样背景下使用用户定义的探针来位点特异性标记蛋白质的通用方法存在迫切需求。
涉及四嗪的逆电子需求狄尔斯-阿尔德(Diels-Alder)反应已显现为快速生物正交反应的重要类别。一些此类反应报告的速率是非常快的。
Yu等人在2012年(Angew.Chem.Int.Ed.第51卷,第10600-10604页)公开了遗传编码的环丙烯在哺乳动物细胞中引导快速、光点击(Photoclick)化学介导的蛋白质标记。作者报告了它在稳定环丙烯氨基酸的合成、在与两种四唑的光诱导环加成反应中的反应性表征、在大肠杆菌(E.coli)中和在哺乳动物细胞中将它位点特异性引入蛋白质,以及它在体外和体内引导蛋白质生物正交(bioothogonal)标记的用途。为了将他们的含环丙烯的氨基酸引入蛋白质,作者不得不开发正交tRNA/tRNA合成酶对,该正交tRNA/tRNA合成酶对响应于TAG琥珀密码子,选择性装载他们的环丙烯赖氨酸氨基酸。这需要构建合成酶文库、使该合成酶内的五个位置随机化、连同至少五轮正向和反向选择筛选。此工作的缺点是它依赖于产生的特定突变型合成酶。在将他们的四唑化合物接合至他们的修饰氨基酸中的环丙烯部分中时,Yu等人使用了光激活。光激活在302纳米或365纳米下进行。在Yu等人的将四唑接合至氨基酸中对光激活的要求是本技术中的缺点。这在缀合化学中是费力的额外步骤。紫外线(UV)还损伤细胞,并且因此在体内/细胞背景中是不利的。
Kamber等人公开了异构环丙烯展示独特的生物正交反应性(2013JACS第135卷,第13680-13683页)。作者论述了可用于在复杂环境中加标签于生物分子的两种反应:四嗪与1,3-二取代环丙烯的逆电子需求Diels-Alder反应,以及腈亚胺与3,3-二取代环丙烯的1,3-偶极环加成。作者论述了用于产生稳定环加合物的各种化学反应方案。由Kamber等人论述的分子都不是氨基酸。没有理由设想可将所述化合物引入氨基酸。即使尝试了任何此类引入,也绝对不存在可能允许将此类化合物引入多肽的建议或指导。Kamber等人未呈现包含所述化学基团中任一种的氨基酸的合成方案。在此文献中的任何地方都未提及用于引入蛋白质的生化工具。Kamber等人只涉及环丙烯上取代模式的检验,一种此类模式允许与四嗪反应,并且一种此类模式不容许与四嗪反应。
本发明试图克服与现有技术相关的一个或多个问题。
技术实现要素:
在一个方面中,本发明提供一种包含含有环丙烯基的氨基酸的多肽,其中将所述环丙烯基经由氨基甲酸酯基接合至氨基酸。
适合地,所述环丙烯基是1,3-二取代环丙烯。适合地,所述环丙烯是1,3-二甲基环丙烯。适合地,所述环丙烯基作为赖氨酸氨基酸的残基存在。适合地,所述多肽进一步包含连接至所述环丙烯基的四嗪化合物。
在另一个方面中,本发明涉及一种包含环丙烯的氨基酸,其中将所述环丙烯基经由氨基甲酸酯基接合至氨基酸部分。
适合地,所述环丙烯是1,3-二取代环丙烯。适合地,所述环丙烯是1,3-二甲基环丙烯。适合地,所述氨基酸是赖氨酸氨基酸。适合地,所述氨基酸包括Nε-[((2-甲基环丙-2-烯-1-基)甲氧基)羰基]-l-赖氨酸。
适合地,所述氨基酸包括下者,或更适合地由下者组成:
在另一个方面中,本发明涉及一种产生包含环丙烯基的多肽的方法,其中将所述环丙烯基经由氨基甲酸酯基接合至氨基酸部分,所述方法包括将氨基酸遗传引入多肽,该氨基酸包含经由氨基甲酸酯基接合至氨基酸部分的环丙烯基。
适合地,产生多肽的步骤包括:
(i)提供编码多肽的核酸,其中核酸包含编码含有环丙烯基的氨基酸的正交密码子;
(ii)在正交tRNA合成酶/tRNA对的存在下翻译所述核酸,该正交tRNA合成酶/tRNA对能够识别所述正交密码子并且将含有环丙烯基的所述氨基酸引入多肽链。
适合地,所述正交密码子包括琥珀密码子(TAG),所述tRNA包括MbtRNACUA,并且所述tRNA合成酶包括MbPylRS。
适合地,所述正交密码子包括琥珀密码子(TAG),所述tRNA包括MmtRNACUA,并且所述tRNA合成酶包括MmPylRS。
在另一个方面中,本发明涉及一种如上述的方法,其中包含环丙烯基的所述氨基酸是如上述的氨基酸。
在另一个方面中,本发明涉及一种产生包含四嗪基的多肽的方法,所述方法包括提供如上述的包含环丙烯基的多肽,使所述多肽与四嗪化合物接触,并且孵育以允许通过逆电子需求狄尔斯-阿尔德环加成反应来将四嗪接合至环丙烯基。
适合地,允许所述反应进行10分钟或更短时间,优选进行1分钟或更短时间,更优选进行30秒或更短时间。反应发生在体内,或在真核培养条件如组织培养基或其他适合于真核细胞的培养基中时,该反应可能需要进行长于30秒以实现最大化标记。熟练操作人员可基于本文提供的指导通过试错法来确定最佳反应时间。
在另一个方面中,本发明涉及一种如上述的多肽,其中所述多肽包含各含有环丙烯基的两个或更多个氨基酸,其中将各所述环丙烯基经由氨基甲酸酯基接合至各所述氨基酸。在多肽上两个或更多个环丙烯基的提供有利地允许将两个或更多个缀合基团(官能团)接合至多肽。当缀合基团(官能团)包含药物分子(如细胞毒分子)如在抗体-药物缀合物中时,这尤其有帮助。
适合地,所述多肽包含各含有环丙烯基的四个氨基酸。
适合地,抗体药物缀合物(ADC)包含如上述的包含各含有环丙烯基的四个氨基酸的多肽。这尤其有利于将四个细胞毒分子接合至目标ADC。
在另一个方面中,本发明涉及一种包含如上述的多肽的抗体药物缀合物(ADC)。适合地,多肽是抗体多肽,如全抗体(例如单克隆抗体(mAb))或者是抗体片段(例如单链可变片段[scFv]),适合地,抗体片段包含CDR氨基酸序列。
适合地,抗体多肽(或片段)可有利地通过一种或多种嵌合抗体多肽的制造来人源化;适合地,抗体多肽(或片段)可有利地是移植CDR的;适合地,抗体多肽(或片段)可有利地被完全人源化至技术容许的程度。
适合地,可将抗体多肽(或片段)融合至另一目标多肽,如转铁蛋白受体的配体(例如转铁蛋白或它的一部分),以协助ADC的运输和/或寻靶。
在另一个方面中,本发明涉及一种如上述的多肽,其中将所述四嗪基进一步接合至荧光团。
适合地,所述荧光团包括荧光素、四甲基罗丹明(TAMRA)或硼-二吡咯亚甲基(BODIPY)。
适合地,所述荧光团可包括一个或多个一种或多种Alexa荧光团。适合地,所述荧光团可包括一个或多个菁基荧光团。
发明详述
遗传密码扩展方法允许具有多样化学结构且带有多样官能团的非天然氨基酸的定量、位点特异性和遗传引导的引入。这最常通过响应于引入目标基因的琥珀终止密码子而插入非天然氨基酸来实现12,13。经由将正交氨酰-tRNA合成酶/tRNACUA对引入细胞来实现遗传密码扩展。吡咯酰-tRNA合成酶/tRNACUA对在最有用的用于遗传密码扩展的配对之中13,因为它:1)可特异性识别一套有用的非天然氨基酸,2)可被开发以识别扩大的一套化学结构,并且3)可在大肠杆菌14、酵母15、哺乳动物细胞16-18、秀丽隐杆线虫(C.elegans)19和黑腹果蝇(D.melanogaster)20中用作用于遗传密码扩展的正交配对。
我们证实具有环丙烯基的新合成蛋白质的产生,可用经由化学选择性逆电子需求狄尔斯-阿尔德反应引入的四嗪探针来标记环丙烯基。
在另一个方面中,本发明涉及一种如上述的同源重组多肽。适合地,通过如上述的方法来制得所述多肽。
还公开了一种根据本文所述的一种或多种方法产生的多肽。以及作为那些新方法的产物,此类多肽具有包含环丙烯(适合地为联接氨基甲酸酯的环丙烯)的技术特征。
突变有它在本领域中的标准意思,并且可指提到的残基、基序或域的取代或截短或删除。可在多肽水平上实现突变,例如通过合成含有突变序列的多肽,或可在核苷酸水平上实现,例如通过制备编码突变序列的核酸,可随后翻译该核酸以产生突变多肽。其中无氨基酸被指定为对给定突变位点的替换氨基酸,适合地,使用随机化的所述位点。作为默认(default)突变,可使用丙氨酸(A)。适合地,在一个或多个特定位点使用的突变如本文所陈述。
片段在长度上适合为至少10个氨基酸,适合为至少25个氨基酸,适合为至少50个氨基酸,适合为至少100个氨基酸,适合为至少200个氨基酸,适合为至少250个氨基酸,适合为至少300个氨基酸,适合为至少313个氨基酸,或适合为目标多肽的大多数。
可在体内或体外实施本发明所述方法。
在一个实施方案中,适合地,本发明的方法未应用于人体或动物体。适合地,本发明所述方法是体外方法。适合地,所述方法不需要人体或动物体的存在。适合地,所述方法不是人体或动物体的诊断或手术或治疗的方法。
术语‘包含’应理解成具有它在本领域中的标准意思,即,包括规定的特征或特征组,但该术语不排除也存在的任何其他规定特征或特征组。
优点
与已知的蛋白质标记基团相比,环丙烯是富碳较少的基团。
当前发明的环丙烯氨基酸导致与现有技术相比更快速的蛋白质标记。
使用本发明的环丙烯氨基酸导致与现有技术标记的氨基酸相比更有效的引入。
已知将带有降冰片烯基的氨基酸引入蛋白质。本发明提供优于涉及降冰片烯基的现有技术方法的特定优点。例如,尽管本发明的环丙烯氨基酸的缀合化学性质与含降冰片烯的氨基酸的相似,但至环丙烯氨基酸的缀合可更快。
根据本发明的环丙烯氨基酸的引入与现有技术非天然氨基酸的引入相比可更有效。根据本发明的环丙烯氨基酸的引入能导致与现有技术非天然氨基酸相比更高水平的引入。
本发明的优点在于可使用野生型tRNA合成酶来引入教导的环丙烯氨基酸。现有技术非天然氨基酸(例如像引入BCN基的氨基酸)趋向于它们的引入需要突变型tRNA合成酶。
将非天然氨基酸引入多肽的快速缀合反应已在现有技术中提及。例如,TCO/BCN氨基酸提供快速的反应时间,这与降冰片烯反应时间相比更快。然而,环丙烯氨基酸的优点在于提供非常快速的反应时间。
某些已知非天然氨基酸能够使用野生型tRNA合成酶。例如,可使用野生型tRNA合成酶来引入包含降冰片烯基的氨基酸。然而,通过使用本发明的含环丙烯的氨基酸可实现更高水平的引入。换言之,与使用现有技术非天然氨基酸如包含降冰片烯的氨基酸时相比,当使用本发明的含环丙烯氨基酸时产生物质(包含非天然氨基酸)的量更大。
本发明的优点在于环丙烯氨基酸形成本文指出的tRNA合成酶的极佳底物,最适合地为本文指出的野生型tRNA合成酶。
本发明的优点在于含环丙烯氨基酸支持极佳的接头化学性质,例如与含四嗪化合物的快速和特异性反应。
本发明的优点在于与先前用于标记蛋白质的已知非天然氨基酸相比,含环丙烯氨基酸的尺寸较小。例如,可将包含降冰片烯的已知非天然氨基酸引入多肽,而本发明的含环丙烯氨基酸与现有技术的含降冰片烯氨基酸相比,有利地具有较小的尺寸。
本发明的优点在于当被引入多肽时,环丙烯氨基酸扰动蛋白质结构的可能性较小。至少部分此有利效应可归因于环丙烯分子基团的小尺寸。
环丙烯基引入的关键优点在于它容许将一套极有用的另外的化合物(如标签)简便和特异性附接至环丙烯基。
在另一个方面中,本发明涉及一种如上述的多肽,其中将所述环丙烯基接合至四嗪基。
环丙烯-氨基甲酸酯连接
包含酰胺键合的环丙烯的非天然氨基酸已描述于现有技术中(Yu等人2012)。此氨基酸是3,3二取代的。此氨基酸如下:
为了将此氨基酸引入多肽,使用突变型tRNA合成酶是必需的。
相反地,本发明的包含环丙烯的氨基酸含有氨基甲酸酯基(而不是酰胺基)。因此,本发明的含环丙烯氨基酸在化学上与本领域酰胺键合的环丙烯氨基酸不同。
本发明的示例性氨基酸是1,3二取代的。本发明的示例性氨基酸如下:
本发明的氨基甲酸酯-环丙烯氨基酸的优点在于它可通过野生型tRNA合成酶很好地引入。这具有需要较少生物操纵来得到良好引入的优点。。这还提供增强或增加的引入的优点。换言之,与已知非天然氨基酸相比,本发明的环丙烯-氨基甲酸酯氨基酸被更高水平和/或更有效地引入。
本发明的环丙烯氨基酸的使用可提供与四嗪化合物反应的优良速率。
本发明的氨基甲酸酯化学性质提供在引入的氨基酸的化学结构中更多自由度的优点。详细而言,与本领域已知的酰胺环丙烯比较,本发明的环丙烯氨基甲酸酯具有更多自由度。类似地,当存在于多肽链中时,本发明的环丙烯氨基甲酸酯更可及。
通过与本领域已知的酰胺键合的环丙烯比较,本发明的环丙烯氨基甲酸酯是轻微“更长的”氨基酸。这提供对于远离氨基酸骨架的氨基酸基团更多“到达”的优点。再次地,这能使那些基团对另外的标记或缀合反应更可及。
本发明的环丙烯氨基甲酸酯的化学结构有利地提供更多构象自由度。换言之,与现有技术的酰胺键合的环丙烯氨基酸相比,本发明的环丙烯氨基甲酸酯基能在蛋白质结构内采取更多构象。
更详细而言,这可起因于环丙烯基与氨基酸基团之间键合的性质。在现有技术的酰胺布置中,重要键是SP2杂化的。在本发明中,重要键是SP3杂化的,这是更灵活的键布置。
此外,本发明的环丙烯氨基甲酸酯布置包含在氨基甲酸酯与环丙烯基之间的亚甲基。首先,这提供更长的分子。现有技术的酰胺键合的版本是不太有利的更短分子。更具体而言,本发明的氨基酸中的亚甲基碳对应于双键合氧基(=o),而不是本发明所述的有利的亚甲基碳。现有技术的酰胺氨基酸中的双键合形式不能和本发明的氨基酸中亚甲基碳键合的基团一样自由旋转。
本发明的氨基酸与现有技术的含降冰片烯的氨基酸相比较小且仍保留有利的氨基甲酸酯化学性质的事实是本发明的益处。此益处尤其提供将氨基酸更好地引入多肽链。
另外,本发明的氨基酸中的环丙烯氨基甲酸酯布置有利地促进至四嗪化合物的接合(四嗪缀合)。
适合地,将所述四嗪基进一步接合至荧光团。
适合地,将所述四嗪基进一步接合至聚乙二醇(PEG)基团。
适合地,所述荧光团包括荧光素、四甲基罗丹明(TAMRA)或硼-二吡咯亚甲基(BODIPY)。
引入
适合地,使用野生型tRNA合成酶来将本发明的环丙烯氨基酸引入多肽。
适合地,在对应于野生型多肽中的赖氨酸残基的位置引入含有环丙烯基的氨基酸。这具有维持含环丙烯的多肽与衍生它的野生型多肽最接近的可能结构联系的优点。
适合地,多肽包含单个环丙烯基。这具有维持对任何另外的化学修饰(可针对环丙烯基)的特异性的优点。例如,当目标多肽中仅存在单个环丙烯基时,那么可有利地避免部分修饰的可能问题(例如,其中多肽中仅一个亚组的环丙烯基被随后修饰),或相同多肽中在交替的环丙烯基之间变化的反应微环境的问题(这能导致多肽中不同定位处不同的一个或多个环丙烯基之间不平等的反应性)。
适合地,多肽包含两个环丙烯基;适合地,多肽包含三个环丙烯基;适合地,多肽包含四个环丙烯基;适合地,多肽包含五个环丙烯基;适合地,多肽包含十个环丙烯基或甚至更多个环丙烯基。
原则上,可通过相同的或通过不同的正交密码子/正交tRNA对来引入多个含环丙烯氨基酸。适合地,通过插入多个琥珀密码子(连同如本文所述的合适的正交tRNA合成酶)来引入多个含环丙烯氨基酸。
适合地,包含环丙烯的氨基酸是赖氨酸氨基酸。在一个实施方案中,tRNA可能来自一个物种,如巴氏甲烷八叠球菌(Methanosarcina barkeri),并且tRNA合成酶可来自另一个物种,如马氏甲烷八叠球菌(Methanosarcina mazei)。在另一个实施方案中,tRNA可来自第一物种,如马氏甲烷八叠球菌,并且tRNA合成酶可来自第二物种,如巴氏甲烷八叠球菌。当正交对包含来自不同物种的tRNA和tRNA合成酶时,总是有正交对能一起有效工作的限制条件,即,tRNA合成酶将有效氨基酰化目标氨基酸的tRNA。同样地,可使用突变型tRNA或突变型tRNA合成酶,前提是它们具有正确的氨基酰化活性。尽管本发明的优点在于使用野生型PylRS合成酶将本发明的含环丙烯氨基酸有效地装载至tRNA上,但同样可能使用突变型PylRS合成酶,前提是它们在用本发明的含环丙烯氨基酸装载tRNA中有效。最适合地,正交对包含来自相同物种的tRNA和tRNA合成酶。
毫无疑问,可能开发野生型合成酶(或另一合适的合成酶变体)来制备用于本发明的环丙烯氨基酸引入的合成酶,该合成酶可能具有提高的效率。原则上,Pyl衍生的tRNA合成酶可能是有用的。可能产生嵌合tRNA合成酶,前提是tRNA合成酶分子的装载/乙酰化部分基于或衍生自Pyl tRNA合成酶。换言之,tRNA分子的反密码子部分可根据操作人员的选择而变化,例如变化为引导tRNA识别交替密码子,如有义密码子、四联体密码子、琥珀密码子或另一“终止”密码子。然而,应保存tRNA分子的有功能的酰化/装载部分,以便保留环丙烯装载活性。
巴氏甲烷八叠球菌和马氏甲烷八叠球菌物种的吡咯赖氨酸tRNA合成酶中的任何一种都是合适的。
巴氏甲烷八叠球菌和马氏甲烷八叠球菌tRNA两者都是合适的。在任何情况下,这些tRNA相差仅一个核苷酸。这种一个核苷酸的差异不影响它们与含环丙烯氨基酸连接的活性。因此,任何一个tRNA在本发明中可同样适用。
使用的tRNA可变化,如经突变。在所有情况下,任何此类Pyl tRNA的变体或突变型应总是保持与用于用含环丙烯氨基酸装载tRNA的tRNA合成酶有成效地相互作用的能力。
遗传引入和多肽产生
在根据本发明的方法中,所述遗传引入优选使用正交或扩展的遗传密码,其中已分配一个或多个特定正交密码子来编码具有环丙烯基的特定氨基酸残基,从而使得可通过使用正交tRNA合成酶/tRNA对来遗传引入它。正交tRNA合成酶/tRNA对原则上可以是任何能够用包含环丙烯基的氨基酸来装载tRNA、并且能够响应于正交密码子将包含环丙烯基的氨基酸引入多肽链的此类对。
正交密码子可能是正交密码子琥珀密码子、赭石密码子、卵白石(opal)密码子或四联体密码子。密码子仅仅必须对应于将用于携带包含环丙烯基的氨基酸的正交tRNA。优选地,正交密码子是琥珀密码子。
应注意,本文示出的许多特定实例使用琥珀密码子和对应的tRNA/tRNA合成酶。如上文指出的,这些可以是变化的。或者,为了在不费力使用或选择能够用包含环丙烯基的氨基酸工作的备选tRNA/tRNA合成酶对的情况下使用其他密码子,仅仅可用所选密码子所期望的反密码子区域调换tRNA的反密码子区域。反密码子区域不涉及tRNA的装载或引入功能,也不由tRNA合成酶识别,因此此类调换完全在熟练操作人员的范围内。因此,在一些实施方案中,用于本发明中的tRNA反密码子区域如MbtRNACUA或MmtRNACUA可互换,即,可使用嵌合tRNACUA以使得反密码子区域被调换来识别交替密码子,从而使得可响应于如本文论述的不同正交密码子(包括赭石密码子、卵白石密码子或四联体密码子)来引入含环丙烯氨基酸,并且编码待引入环丙烯氨基酸的多肽的核酸经对应地突变以在环丙烯氨基酸的引入点引入同族密码子。最适合地,正交密码子是琥珀密码子。
因此,需要时可使用备选正交tRNA合成酶/tRNA对。
优选地,正交合成酶/tRNA对是巴氏甲烷八叠球菌MS吡咯赖氨酸tRNA合成酶(MbPylRS)和它的同族琥珀抑制子tRNA(MbtRNACUA)。
巴氏甲烷八叠球菌PylT基因编码MbtRNACUA tRNA。
巴氏甲烷八叠球菌PylS基因编码MbPylRS tRNA合成酶蛋白。当使用数字地址来提及特定氨基酸残基时,使用MbPylRS(巴氏甲烷八叠球菌吡咯酰-tRNA合成酶)氨基酸序列作为参考序列(即,如由可公开得到的野生型巴氏甲烷八叠球菌PylS基因登录号Q46E77编码的)来获得编号:
MDKKPLDVLI SATGLWMSRT GTLHKIKHYE VSRSKIYIEM ACGDHLVVNN
SRSCRTARAF RHHKYRKTCK RCRVSDEDIN NFLTRSTEGK TSVKVKVVSA
PKVKKAMPKS VSRAPKPLEN PVSAKASTDT SRSVPSPAKS TPNSPVPTSA
PAPSLTRSQL DRVEALLSPE DKISLNIAKP FRELESELVT RRKNDFQRLY
TNDREDYLGK LERDITKFFV DRDFLEIKSP ILIPAEYVER MGINNDTELS
KQIFRVDKNL CLRPMLAPTL YNYLRKLDRI LPDPIKIFEV GPCYRKESDG
KEHLEEFTMV NFCQMGSGCT RENLESLIKE FLDYLEIDFE IVGDSCMVYG
DTLDIMHGDL ELSSAVVGPV PLDREWGIDK PWIGAGFGLE RLLKVMHGFK
NIKRASRSES YYNGISTNL。
如果有需要,本领域技术人员可通过突变MbPylRS tRNA合成酶来修改它,从而使其针对待使用的环丙烯氨基酸而优化。对突变的需求(如果存在的话)取决于使用的环丙烯氨基酸。在一个实例中,当环丙烯氨基酸未由MbPylRS tRNA合成酶蛋白加工时,MbPylRS tRNA合成酶可能需要进行突变。
此类突变(需要时)可通过将突变引入MbPylRS tRNA合成酶来进行,例如在MbPylRS tRNA合成酶中的一个或多个以下位置:M241、A267、Y271、L274和C313。
tRNA合成酶
本发明的tRNA合成酶可变化。尽管在实例中可能已使用特定tRNA合成酶序列,但本发明不意图仅限制于那些实例。
原则上,在本发明中可使用任何提供相同tRNA装载(氨酰化)功能的tRNA合成酶。
例如,tRNA合成酶可来自任何合适的物种,如来自古菌域,例如来自巴氏甲烷八叠球菌(Methanosarcina barkeri)MS;巴氏甲烷八叠球菌str.Fusaro;马氏甲烷八叠球菌(Methanosarcina mazei)Go1;嗜乙酸甲烷八叠球菌(Methanosarcina acetivorans)C2A;嗜热甲烷八叠球菌(Methanosarcina thermophila);或布氏拟甲烷球菌(Methanococcoides burtonii)。或者,tRNA合成酶可来自细菌,例如来自哥本哈根脱亚硫酸菌(Desulfitobacterium hafniense)DCB-2;哥本哈根脱亚硫酸菌Y51;哥本哈根脱亚硫酸菌PCP1;醋酸氧化脱硫肠状菌(Desulfotomaculum acetoxidans)DSM 771。
来自这些生物的示例性序列是可公开得到的序列。提供以下实例作为吡咯赖氨酸tRNA合成酶的示例性序列:
>M.barkeriMS/1-419/
巴氏甲烷八叠球菌MS
版本Q6WRH6.1GI:74501411
MDKKPLDVLISATGLWMSRTGTLHKIKHHEVSRSKIYIEMACGDHLVVNNSRSCRTARAFRHHKYRKTCKRCRVSDEDINNFLTRSTESKNSVKVRVVSAPKVKKAMPKSVSRAPKPLENSVSAKASTNTSRSVPSPAKSTPNSSVPASAPAPSLTRSQLDRVEALLSPEDKISLNMAKPFRELEPELVTRRKNDFQRLYTNDREDYLGKLERDITKFFVDRGFLEIKSPILIPAEYVERMGINNDTELSKQIFRVDKNLCLRPMLAPTLYNYLRKLDRILPGPIKIFEVGPCYRKESDGKEHLEEFTMVNFCQMGSGCTRENLEALIKEFLDYLEIDFEIVGDSCMVYGDTLDIMHGDLELSSAVVGPVSLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRASRSESYYNGISTNL
>M.barkeriF/1-419/
巴氏甲烷八叠球菌str.Fusaro
版本YP_304395.1GI:73668380
MDKKPLDVLISATGLWMSRTGTLHKIKHYEVSRSKIYIEMACGDHLVVNNSRSCRTARAFRHHKYRKTCKRCRVSDEDINNFLTRSTEGKTSVKVKVVSAPKVKKAMPKSVSRAPKPLENPVSAKASTDTSRSVPSPAKSTPNSPVPTSAPAPSLTRSQLDRVEALLSPEDKISLNIAKPFRELESELVTRRKNDFQRLYTNDREDYLGKLERDITKFFVDRDFLEIKSPILIPAEYVERMGINNDTELSKQIFRVDKNLCLRPMLAPTLYNYLRKLDRILPDPIKIFEVGPCYRKESDGKEHLEEFTMVNFCQMGSGCTRENLESLIKEFLDYLEIDFEIVGDSCMVYGDTLDIMHGDLELSSAVVGPVPLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRASRSESYYNGISTNL
>M.mazei/1-454
马氏甲烷八叠球菌Go1
版本NP_633469.1GI:21227547
MDKKPLNTLISATGLWMSRTGTIHKIKHHEVSRSKIYIEMACGDHLVVNNSRSSRTARALRHHKYRKTCKRCRVSDEDLNKFLTKANEDQTSVKVKVVSAPTRTKKAMPKSVARAPKPLENTEAAQAQPSGSKFSPAIPVSTQESVSVPASVSTSISSISTGATASALVKGNTNPITSMSAPVQASAPALTKSQTDRLEVLLNPKDEISLNSGKPFRELESELLSRRKKDLQQIYAEERENYLGKLEREITRFFVDRGFLEIKSPILIPLEYIERMGIDNDTELSKQIFRVDKNFCLRPMLAPNLYNYLRKLDRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFCQMGSGCTRENLESIITDFLNHLGIDFKIVGDSCMVYGDTLDVMHGDLELSSAVVGPIPLDREWGIDKPWIGAGFGLERLLKVKHDFKNIKRAARSESYYNGISTNL
>M.acetivorans/1-443
嗜乙酸甲烷八叠球菌C2A
版本NP_615128.2GI:161484944
MDKKPLDTLISATGLWMSRTGMIHKIKHHEVSRSKIYIEMACGERLVVNNSRSSRTARALRHHKYRKTCRHCRVSDEDINNFLTKTSEEKTTVKVKVVSAPRVRKAMPKSVARAPKPLEATAQVPLSGSKPAPATPVSAPAQAPAPSTGSASATSASAQRMANSAAAPAAPVPTSAPALTKGQLDRLEGLLSPKDEISLDSEKPFRELESELLSRRKKDLKRIYAEERENYLGKLEREITKFFVDRGFLEIKSPILIPAEYVERMGINSDTELSKQVFRIDKNFCLRPMLAPNLYNYLRKLDRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFCQMGSGCTRENLEAIITEFLNHLGIDFEIIGDSCMVYGNTLDVMHDDLELSSAVVGPVPLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRAARSESYYNGISTNL
>M.thermophila/1-478
嗜热甲烷八叠球菌,版本DQ017250.1GI:67773308
MDKKPLNTLISATGLWMSRTGKLHKIRHHEVSKRKIYIEMECGERLVVNNSRSCRAARALRHHKYRKICKHCRVSDEDLNKFLTRTNEDKSNAKVTVVSAPKIRKVMPKSVARTPKPLENTAPVQTLPSESQPAPTTPISASTTAPASTSTTAPAPASTTAPAPASTTAPASASTTISTSAMPASTSAQGTTKFNYISGGFPRPIPVQASAPALTKSQIDRLQGLLSPKDEISLDSGTPFRKLESELLSRRRKDLKQIYAEEREHYLGKLEREITKFFVDRGFLEIKSPILIPMEYIERMGIDNDKELSKQIFRVDNNFCLRPMLAPNLYNYLRKLNRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFCQMGSGCTRENLEAIIKDFLDYLGIDFEIVGDSCMVYGDTLDVMHGDLELSSAVVGPVPMDRDWGINKPWIGAGFGLERLLKVMHNFKNIKRASRSESYYNGISTNL
>M.burtonii/1-416
布氏拟甲烷球菌DSM 6242,版本YP_566710.1GI:91774018
MEKQLLDVLVELNGVWLSRSGLLHGIRNFEITTKHIHIETDCGARFTVRNSRSSRSARSLRHNKYRKPCKRCRPADEQIDRFVKKTFKEKRQTVSVFSSPKKHVPKKPKVAVIKSFSISTPSPKEASVSNSIPTPSISVVKDEVKVPEVKYTPSQIERLKTLMSPDDKIPIQDELPEFKVLEKELIQRRRDDLKKMYEEDREDRLGKLERDITEFFVDRGFLEIKSPIMIPFEYIERMGIDKDDHLNKQIFRVDESMCLRPMLAPCLYNYLRKLDKVLPDPIRIFEIGPCYRKESDGSSHLEEFTMVNFCQMGSGCTRENMEALIDEFLEHLGIEYEIEADNCMVYGDTIDIMHGDLELSSAVVGPIPLDREWGVNKPWMGAGFGLERLLKVRHNYTNIRRASRSELYYNGINTNL
>D.hafniense_DCB-2/1-279
哥本哈根脱亚硫酸菌DCB-2
版本YP_002461289.1GI:219670854
MSSFWTKVQYQRLKELNASGEQLEMGFSDALSRDRAFQGIEHQLMSQGKRHLEQLRTVKHRPALLELEEGLAKALHQQGFVQVVTPTIITKSALAKMTIGEDHPLFSQVFWLDGKKCLRPMLAPNLYTLWRELERLWDKPIRIFEIGTCYRKESQGAQH LNEFTMLNLTELGTPLEERHQRLEDMARWVLEAAGIREFELVTESSVVYGDTVDVMKGDLELASGAMGPHFLDEKWEIVDPWVGLGFGLERLLMIREGTQHVQSMARSLSYLDGVRLNIN
>D.hafniense_Y51/1-312
哥本哈根脱亚硫酸菌Y51
版本YP_521192.1GI:89897705
MDRIDHTDSKFVQAGETPVLPATFMFLTRRDPPLSSFWTKVQYQRLKELNASGEQLEMGFSDALSRDRAFQGIEHQLMSQGKRHLEQLRTVKHRPALLELEEGLAKALHQQGFVQVVTPTIITKSALAKMTIGEDHPLFSQVFWLDGKKCLRPMLAPNLYTLWRELERLWDKPIRIFEIGTCYRKESQGAQHLNEFTMLNLTELGTPLEERHQRLEDMARWVLEAAGIREFELVTESSVVYGDTVDVMKGDLELASGAMGPHFLDEKWEIVDPWVGLGFGLERLLMIREGTQHVQSMARSLSYLDGVRLNIN
>D.hafniensePCP1/1-288
哥本哈根脱亚硫酸菌
版本AY692340.1GI:53771772
MFLTRRDPPLSSFWTKVQYQRLKELNASGEQLEMGFSDALSRDRAFQGIEHQLMSQGKRHLEQLRTVKHRPALLELEEKLAKALHQQGFVQVVTPTIITKSALAKMTIGEDHPLFSQVFWLDGKKCLRPMLAPNLYTLWRELERLWDKPIRIFEIGTCYRKESQGAQHLNEFTMLNLTELGTPLEERHQRLEDMARWVLEAAGIREFELVTESSVVYGDTVDVMKGDLELASGAMGPHFLDEKWEIFDPWVGLGFGLERLLMIREGTQHVQSMARSLSYLDGVRLNIN
>D.acetoxidans/1-277
醋酸氧化脱硫肠状菌DSM 771
版本YP_003189614.1GI:258513392
MSFLWTVSQQKRLSELNASEEEKNMSFSSTSDREAAYKRVEMRLINESKQRLNKLRHETRPAICALENRLAAALRGAGFVQVATPVILSKKLLGKMTITDEHALFSQVFWIEENKCLRPMLAPNLYYILKDLLRLWEKPVRIFEIGSCFRKESQGSNHLNEFTMLNLVEWGLPEEQRQKRISELAKLVMDETGIDEYHLEHAESVVYGETVDVMHRDIELGSGALGPHFLDGRWGVVGPWVGIGFGLERLLMVEQGGQNVRSMGKSLTYLDGVRLNI
当已通过突变tRNA合成酶来提供特定tRNA装载(氨酰化)功能时,那么可能仅仅使用另一野生型tRNA序列(例如选自上述的序列)是不适当的。在这种情境下,保留相同的tRNA装载(氨酰化)功能将是重要的。这是通过将示例性tRNA合成酶中的一个或多个突变移入替代的(alternate)tRNA合成酶骨架(如选自上述的一个)来完成的。
以这种方式,应可能将选定的突变移至对应tRNA合成酶序列,如来自除示例性M.barkeri和/或M.mazei序列以外的其他生物的对应pylS序列。
可通过与已知tRNA合成酶(如示例性M.barkeri和/或M.mazei序列)比对来选择靶tRNA合成酶蛋白质/骨架。
现在参考pylS(吡咯赖氨酸tRNA合成酶)序列来说明此主题,但所述原理同样应用于特定目标tRNA合成酶。
例如,可准备所有PylS序列的比对。这些可具有低总%序列同一性。因此,如通过使序列与已知tRNA合成酶比对(而不是仅仅使用低序列同一性得分)来研究序列是重要的,以确保使用的序列确实是tRNA合成酶。
因此,适合地,当考虑到序列同一性时,适合地,在上述tRNA合成酶实例的序列间考虑。适合地,%同一性可如根据上述序列的比对而定义。
聚焦于催化区域可能是有用的。此目的在于提供tRNA催化区域,根据其可限定高%同一性,以捕获/鉴定适合于接受为了产生相同的tRNA装载(氨酰化)功能而移植的突变的骨架支架,例如新的或非天然氨基酸识别。
因此,适合地,当考虑到序列同一性时,适合地,在催化区域间考虑。适合地,%同一性可根据催化区域而定义。
可通过编码tRNA合成酶骨架的核苷酸序列的定点诱变来‘转移’或‘移植’突变至替代的tRNA合成酶骨架上。此技术是本领域众所周知的。基本上,骨架pylS序列被选定(例如使用上述活性位点比对),并且将选定的突变转移至(即,在其中制得)对应/同源位置。
当使用数字地址提及特殊氨基酸残基时,除非以其他方式显而易见,否则使用MbPylRS(巴氏甲烷八叠球菌吡咯酰-tRNA合成酶)氨基酸序列作为参考序列(即,如由可公开得到的野生型巴氏甲烷八叠球菌PylS基因登录号Q46E77编码的)来进行编号:
MDKKPLDVLI SATGLWMSRT GTLHKIKHYE VSRSKIYIEM ACGDHLVVNN
SRSCRTARAF RHHKYRKTCK RCRVSDEDIN NFLTRSTEGK TSVKVKVVSA
PKVKKAMPKS VSRAPKPLEN PVSAKASTDT SRSVPSPAKS TPNSPVPTSA
PAPSLTRSQL DRVEALLSPE DKISLNIAKP FRELESELVT RRKNDFQRLY
TNDREDYLGK LERDITKFFV DRDFLEIKSP ILIPAEYVER MGINNDTELS
KQIFRVDKNL CLRPMLAPTL YNYLRKLDRI LPDPIKIFEV GPCYRKESDG
KEHLEEFTMV NFCQMGSGCT RENLESLIKE FLDYLEIDFE IVGDSCMVYG
DTLDIMHGDL ELSSAVVGPV PLDREWGIDK PWIGAGFGLE RLLKVMHGFK
NIKRASRSES YYNGISTNL
如本领域熟知的,这用于定位目标残基。这并非总是严格的计数运用——必须注意背景或比对。例如,如果目标蛋白质的长度轻微不同,则在该序列中对应于(例如)L266的正确残基的位置可能需要比对序列并且挑选等价或对应残基,而不是仅仅采用目标序列的第266位残基。这很好地在有经验读者的能力范围内。
本文使用的突变符号是本领域中的标准。例如,L266M意指用M替换在野生型序列位置266处对应于L的氨基酸。
现在参考示例性M.barkeri和M.mazei序列说明替代的tRNA骨架之间的突变移植,但相同的原理同样应用于至其他骨架上或来自其他骨架的移植。
例如,Mb AcKRS是用于AcK引入的工程化合成酶
亲本蛋白质/骨架:M.barkeri PylS
突变:L266V、L270I、Y271F、L274A、C317F
Mb PCKRS:用于PCK引入的工程化合成酶
亲本蛋白质/骨架:M.barkeri PylS
突变:M241F、A267S、Y271C、L274M
可通过将这些突变移入M.mazei PylS来得到具有相同底物特异性的合成酶。因此,可通过将来自Mb骨架的突变移植至Mm tRNA骨架上来产生以下合成酶:
Mm AcKRS将突变L301V、L305I、Y306F、L309A、C348F引入M.mazei PylS,以及
Mm PCKRS将突变M276F、A302S、Y306C、L309M引入M.mazei PylS。
在下方给出这些示例性移植突变的合成酶的全长序列。
>Mb_PylS/1-419
MDKKPLDVLISATGLWMSRTGTLHKIKHHEVSRSKIYIEMACGDHLVVNNSRSCRTARAFRHHKYRKTCKRCRVSDEDINNFLTRSTESKNSVKVRVVSAPKVKKAMPKSVSRAPKPLENSVSAKASTNTSRSVPSPAKSTPNSSVPASAPAPSLTRSQLDRVEALLSPEDKISLNMAKPFRELEPELVTRRKNDFQRLYTNDREDYLGKLERDITKFFVDRGFLEIKSPILIPAEYVERMGINNDTELSKQIFRVDKNLCLRPMLAPTLYNYLRKLDRILPGPIKIFEVGPCYRKESDGKEHLEEFTMVNFCQMGSGCTRENLEALIKEFLDYLEIDFEIVGDSCMVYGDTLDIMHGDLELSSAVVGPVSLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRASRSESYYNGISTNL
>Mb_AcKRS/1-419
MDKKPLDVLISATGLWMSRTGTLHKIKHHEVSRSKIYIEMACGDHLVVNNSRSCRTARAFRHHKYRKTCKRCRVSGEDINNFLTRSTESKNSVKVRVVSAPKVKKAMPKSVSRAPKPLENSVSAKASTNTSRSVPSPAKSTPNSSVPASAPAPSLTRSQLDRVEALLSPEDKISLNMAKPFRELEPELVTRRKNDFQRLYTNDREDYLGKLERDITKFFVDRGFLEIKSPILIPAEYVERMGINNDTELSKQIFRVDKNLCLRPMVAPTIFNYARKLDRILPGPIKIFEVGPCYRKESDGKEHLEEFTMVNFFQMGSGCTRENLEALIKEFLDYLEIDFEIVGDSCMVYGDTLDIMHGDLELSSAVVGPVSLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRASRSESYYNGISTNL
>Mb_PCKRS/1-419
MDKKPLDVLISATGLWMSRTGTLHKIKHHEVSRSKIYIEMACGDHLVVNNSRSCRTARAFRHHKYRKTCKRCRVSDEDINNFLTRSTESKNSVKVRVVSAPKVKKAMPKSVSRAPKPLENSVSAKASTNTSRSVPSPAKSTPNSSVPASAPAPSLTRSQLDRVEALLSPEDKISLNMAKPFRELEPELVTRRKNDFQRLYTNDREDYLGKLERDITKFFVDRGFLEIKSPILIPAEYVERFGINNDTELSKQIFRVDKNLCLRPMLSPTLCNYMRKLDRILPGPIKIFEVGPCYRKESDGKEHLEEFTMVNFCQMGSGCTRENLEALIKEFLDYLEIDFEIVGDSCMVYGDTLDIMHGDLELSSAVVGPVSLDREWGIDKPWIGAGFGLERLLKVMHGFKNIKRASRSESYYNGISTNL
>Mm_PylS/1-454
MDKKPLNTLISATGLWMSRTGTIHKIKHHEVSRSKIYIEMACGDHLVVNNSRSSRTARALRHHKYRKTCKRCRVSDEDLNKFLTKANEDQTSVKVKVVSAPTRTKKAMPKSVARAPKPLENTEAAQAQPSGSKFSPAIPVSTQESVSVPASVSTSISSISTGATASALVKGNTNPITSMSAPVQASAPALTKSQTDRLEVLLNPKDEISLNSGKPFRELESELLSRRKKDLQQIYAEERENYLGKLEREITRFFVDRGFLEIKSPILIPLEYIERMGIDNDTELSKQIFRVDKNFCLRPMLAPNLYNYLRKLDRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFCQMGSGCTRENLESIITDFLNHLGIDFKIVGDSCMVYGDTLDVMHGDLELSSAVVGPIPLDREWGIDKPWIGAGFGLERLLKVKHDFKNIKRAARSESYYNGISTNL
>Mm_AcKRS/1-454
MDKKPLNTLISATGLWMSRTGTIHKIKHHEVSRSKIYIEMACGDHLVVNNSRSSRTARALRHHKYRKTCKRCRVSDEDLNKFLTKANEDQTSVKVKVVSAPTRTKKAMPKSVARAPKPLENTEAAQAQPSGSKFSPAIPVSTQESVSVPASVSTSISSISTGATASALVKGNTNPITSMSAPVQASAPALTKSQTDRLEVLLNPKDEISLNSGKPFRELESELLSRRKKDLQQIYAEERENYLGKLEREITRFFVDRGFLEIKSPILIPLEYIERMGIDNDTELSKQIFRVDKNFCLRPMVAPNIFNYARKLDRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFFQMGSGCTRENLESIITDFLNHLGIDFKIVGDSCMVYGDTLDVMHGDLELSSAVVGPIPLDREWGIDKPWIGAGFGLERLLKVKHDFKNIKRAARSESYYNGISTNL
>Mm_PCKRS/1-454
MDKKPLNTLISATGLWMSRTGTIHKIKHHEVSRSKIYIEMACGDHLVVNNSRSSRTARALRHHKYRKTCKRCRVSDEDLNKFLTKANEDQTSVKVKVVSAPTRTKKAMPKSVARAPKPLENTEAAQAQPSGSKFSPAIPVSTQESVSVPASVSTSISSISTGATASALVKGNTNPITSMSAPVQASAPALTKSQTDRLEVLLNPKDEISLNSGKPFRELESELLSRRKKDLQQIYAEERENYLGKLEREITRFFVDRGFLEIKSPILIPLEYIERFGIDNDTELSKQIFRVDKNFCLRPMLSPNLCNYMRKLDRALPDPIKIFEIGPCYRKESDGKEHLEEFTMLNFCQMGSGCTRENLESIITDFLNHLGIDFKIVGDSCMVYGDTLDVMHGDLELSSAVVGPIPLDREWGIDKPWIGAGFGLERLLKVKHDFKNIKRAARSESYYNGISTNL
相同的原理同样应用于其他突变和/或其他骨架。
有利地,应测试以这种方式产生的移植多肽来确保已保留期望的功能/底物特异性。
可将编码用于上述方法的目标多肽的多核苷酸引入重组可复制载体。载体可用于在相容宿主细胞中复制核酸。因此,在一个另外实施方案中,本发明提供一种制备本发明的多核苷酸的方法,通过将本发明的多核苷酸引入可复制载体、将载体引入相容宿主细胞、并且使宿主细胞在引起载体复制的条件下生长来实现该方法。可从宿主细胞回收载体。合适的宿主细胞包括细菌,如大肠杆菌。
优选地,将载体中的本发明的多核苷酸有效地连接至控制序列,该控制序列能够通过宿主细胞供应编码序列的表达,即,载体是表达载体。术语“有效地连接”意指所述组分处在容许它们以其预期方式起作用的关系中。将“有效地连接"至编码序列的调节序列以下述方式连接:使得在与控制序列相容的条件下实现编码序列的表达。
可将本发明的载体转化或转染入如描述用来提供本发明的蛋白质表达的合适的宿主细胞。此方法可包括在提供编码蛋白的编码序列的载体的表达的条件下培养用如上述表达载体转化的宿主细胞,并且任选地回收表达的蛋白质。
载体可以是例如具备复制起点,任选地用于所述多核苷酸表达的启动子以及任选地启动子的调节子的质粒或病毒载体。载体可含有一个或多个可选择标记基因,例如在细菌质粒的情况下含有氨苄青霉素抗性基因。载体可用于(例如)转染或转化宿主细胞。
被有效地连接至编码本发明的蛋白质的序列的控制序列包括启动子/增强子和其他表达调节信号。这些控制序列可被选择以与宿主细胞(表达载体被设计于其中使用)相容。术语启动子是本领域众所周知的,并且包括在尺寸和复杂性上处于一定范围的从最小启动子至包括上游元件和增强子的启动子的核酸区域。
本发明的另一个方面是一种将一个或多个含环丙烯氨基酸遗传地和位点特异性引入所选蛋白质的方法,如体外方法,适合地,在真核细胞中。通过所述方法遗传引入的优点在于它排除了包含环丙烯氨基酸的蛋白质一旦形成后被递送入细胞的需要,因为在此实施方案中,它们可直接合成于靶细胞中。该方法包括以下步骤:
i)在编码蛋白质的核苷酸序列中的期望位点处引入正交密码子,或用正交密码子如琥珀密码子替换特定密码子
ii)在细胞中引入正交tRNA合成酶/tRNA对的表达系统,如吡咯赖氨酰-tRNA合成酶/tRNA对
iii)使细胞在具有根据本发明的含环丙烯氨基酸的培养基中生长。
步骤(i)在蛋白质的遗传序列中的期望位点处需要或用正交密码子如琥珀密码子替换特定密码子。这可以通过仅仅引入具有编码蛋白质的核苷酸序列的构建物(如质粒)来实现,其中将期望引入/替换的含环丙烯氨基酸的位点改变,以包含正交密码子如琥珀密码子。这较好在本领域技术人员的能力范围内,并且在此处下方给出此类实例。
步骤(ii)需要正交表达系统以在期望位置(例如琥珀密码子)处特异性引入含环丙烯氨基酸。因此,需要特定正交tRNA合成酶,如正交吡咯赖氨酰-tRNA合成酶,以及特定的对应的正交tRNA对,二者一起能够用含环丙烯氨基酸装载所述tRNA。本文提供了这些的实例。
蛋白质表达和纯化
包含本发明的多核苷酸的宿主细胞可用于表达本发明的蛋白质。可在允许本发明的蛋白质表达的合适条件下培养宿主细胞。本发明的蛋白质的表达可以是组成型的,以使得蛋白质持续产生;或者是诱导型的,其需要刺激物以引发表达。在诱导型表达的情况下,当被例如诱导剂物质(例如地塞米松或IPTG)添加至培养基所需要时,可引发蛋白质产生。
可通过各种本领域已知技术自宿主细胞提取本发明的蛋白质,包括酶促、化学和/或渗透性裂解以及物理破坏。
可通过本领域已知的标准技术如制备色谱、亲和纯化或任何其他合适的技术来纯化本发明的蛋白质。
另外的优点
Yu等人将四唑接合至多肽中的环丙烯氨基酸。Yu等人需要使用紫外照射以便光激活他们的缀合基团。用302纳米紫外照射实现了他们的最佳反应速率。然而,此类型的紫外照射具有高离子化电位。这意味着照射指向的分子和/或细胞可能被此紫外能量损伤。相比之下,本发明的缀合不需要任何用于光激活的紫外步骤。即使当Yu等人使用紫外照射的较小损伤源(例如365纳米紫外照射)时,观察到的反应速率也与本发明提供的那些相比显著更慢。因此,即使Yu等人调整了紫外照射以试图尝试避免或减少与紫外处理相关的一些缺点,仍必须进行相同的费力的照射步骤并且达到更慢的反应速率。本发明的优点在于可省去紫外照射,并且即使不用光激活也能得到极佳的反应速率。
本发明的环丙烯氨基酸的优点在于它们易于制备。例如,合成途径中步骤数有利地很少。
应注意,Yu等人的现有技术的环丙烯氨基酸含有酰胺基。此酰胺键是肽酶的潜在底物。在现有技术的环丙烯氨基酸的酰胺键上的肽酶作用将从多肽上断开分子的环丙烯部分。这明显是不利的。相比之下,本发明的氨基甲酸酯连接的环丙烯基的优点在于氨基甲酸酯键合的环丙烯不是肽酶的靶标。
基于现有技术的技术依赖四唑的缀合化学性质。相反地,本发明教导有利的四嗪化学性质的使用。
本发明的氨基甲酸酯键合的环丙烯氨基酸的优点在于它们允许野生型PylRS合成酶的使用。使用野生型合成酶是有利的,因为它通过减轻制备突变型合成酶的需要而涉及较少劳动。另外,突变型合成酶不总能在tRNA中氨基酰化至与野生型tRNA合成酶相同的水平。换言之,为了操纵现有技术的环丙烯酰胺键合的氨基酸,需要对合成酶制造突变,该突变可能造成氨基酰化效率的损失。相反地,本文证实,使用野生型合成酶对本发明的氨基酸进行氨基酰化是非常有效的方法,这是超过现有技术的另外的优点。
另外的特定和优选方面陈述于随附的独立和从属权利要求中。从属权利要求的特征可适当地与独立权利要求的特征结合,并且可能以不同于在权利要求中明确列出的其他组合方式来结合。
其中设备特征被描述为有效的,以提供功能的,将理解这包括提供该功能或经调试或配置来提供功能的设备特征。
附图说明
现在将参考附图进一步描述本发明的实施方案,其中:
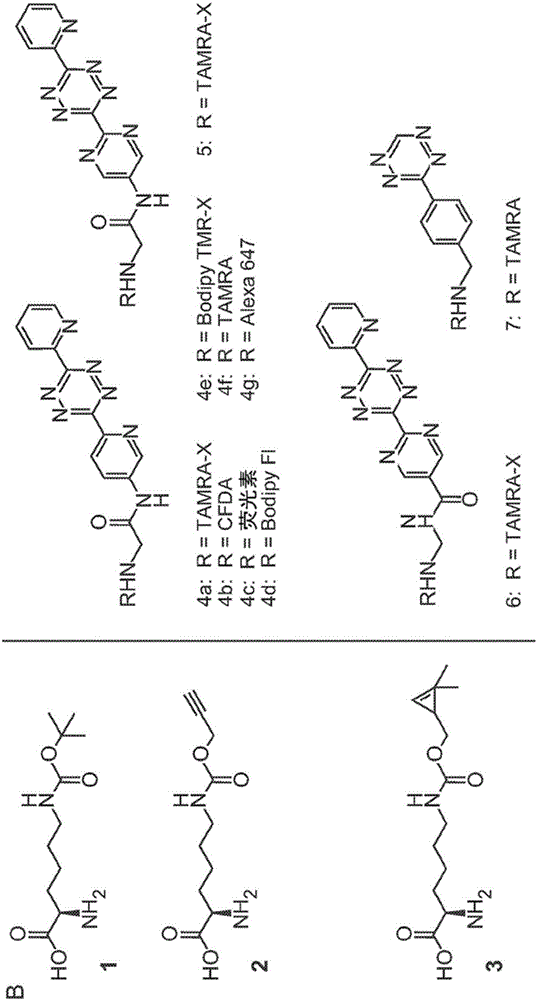
图1 SORT-M允许在多样的密码子处、用多样的化学药品、以及在遗传靶标细胞和组织中的蛋白质组加标签和标记。(a)经由SORT(翻译的随机正交再编码,stochastic orthogonal recoding of translation)的蛋白质组加标签使用正交氨酰-tRNA合成酶/tRNA对。此研究中使用吡咯酰-tRNA合成酶/tRNA对。此合成酶(以及它先前开发的活性位点变体)识别一套非天然氨基酸(黄色星形,以及黄色六边形),不将内源tRNA氨基酰化,但将它的同族tRNA有效氨基酰化——不考虑反密码子同一性;PyltRNA不是内源氨酰-tRNA合成酶的底物。已改变其中反密码子的正交吡咯酰-tRNA合成酶/tRNAXXX对(XXX指示反密码子的选择,黄色)经由与天然合成酶和tRNA(深蓝色和粉红色)所使用以引导天然氨基酸的途径正交的途径来竞争有义密码子(深蓝色和粉红色)的解码。SORT允许响应于多样的密码子将多样的化学基团引入蛋白质组。因为在正交合成酶的活性位点处不存在竞争,所以不需要饥饿和基本培养基。另外,可遗传引导正交蛋白质组加标签系统的表达模式,以允许组织特异性蛋白质组标记。在补充性图1中示出选择压力引入方法,用来与SORT比较。(b)经由3用四嗪探针(4a-g、5、6和7)的SORT和化学选择修饰来在蛋白质组上组合编码氨基酸(1-3),该组合允许经由SORT-M(翻译的随机正交再编码和化学选择修饰,stochastic orthogonal recoding of translation and chemoselective modification)来检测标记的蛋白质。氨基酸结构:Nε-((叔-丁氧基)羰基)-L-赖氨酸 1,Nε-(1-丙炔氧基)羰基)-L-赖氨酸 2(Nε-(1-propynlyoxy)carbonyl)-L-lysine 2)和Nε-(((2-甲基环丙-2-烯-1-基)甲氧基)羰基)-L-赖氨酸。
图2(补充性图2)示出将3定量、位点特异性引入在大肠杆菌中表达的蛋白质,以及它使用四嗪探针的快速和定量标记
A.PylRS/tRNACUA对引导3被有效、位点特异性引入在位置150处带有琥珀终止密码子的sfGFP。3的引入与已良好确立的PylRS/tRNACUA对的极佳底物1相比更有效。
B.用10当量四嗪荧光团4a来特异性和定量标记带有3的2nmol sfGFP。用1mM带有PylRS/tRNACUA对和SfGFP150TAG的3使大肠杆菌生长,自该大肠杆菌纯化的sfGFP-3的ESI-MS分析证实3的引入。sfGFP150-3:预期质量:27951.5Da,实测质量:27950±1.0Da,次峰27820对应于N-端甲硫氨酸的损失。如由标记反应的ESI-MS判断的,用4a标记sfGFP150-3是定量的。预期质量:28758.4Da,实测质量:28758±1.0Da,次峰28627对应于N-端甲硫氨酸的损失。
C.确定用10当量4a标记sfGFP-3(10.6μM,sfGFP在位置150处引入3)的速率常数。用20nmol四嗪-染料缀合物4a(10μl的于DMSO中的2mM溶液)孵育2nmol纯化的sfGFP-3(10.6μM,在20mM Tris-HCl、100mM NaCl、2mM EDTA中,pH 7.4)。在不同时间点自溶液取8μL等分试样,并且将其用700倍过量BCN淬灭且浸没入液氮。用补充有5%β-巯基乙醇的NuPAGE LDS样品缓冲液混合样品,样品被加热10分钟至90℃并且通过4-12%SDS page来分析。通过使用Typhoon Trio磷光影像分析仪(GE Life Sciences)扫描荧光条带来量化标记的蛋白质的量。用ImageQuantTM TL软件(GE Life Sciences)使用橡胶带背景扣除(rubber band background subtraction)来量化条带。通过将数据拟合至单指数方程来测定速率常数。计算观测的速率k'除以4a的浓度得到反应的速率常数k。进行三重测量。使用Kaleidagraph软件(Synergy Software,Reading,UK)执行所有数据处理。为了比较标记带有Nε-5-降冰片烯-2-基氧羰基-L-赖氨酸(NorK,PylRS的已知底物)的sfGFP的速率,使用11.25μM在位置150处带有NorK的sfGFP(SfGFP-NorK)和20当量的4a以相似方式来测定速率。
图3示出补充表1-引物
图4(补充性图3)示出SORT-M允许在大肠杆菌中密码子特异性的蛋白质组加标签和标记
A.使用3经由指示的PylRS/tRNAXXX对来标记蛋白质组。细胞含有两种质粒,一种编码MbPylRS,另一种编码T4溶菌酶和指示的tRNAXXX。在0.1mM 3的存在下从OD600=0.2使细胞生长,并且在1h后通过添加0.2mM阿拉伯糖诱导T4溶菌酶表达。在又3h后收获细胞。经由在引入的3与四嗪荧光团4a之间的逆电子需求狄尔斯-阿尔德反应(20mM,1h,室温)来检测裂解物中的加标签的蛋白质。括号中的氨基酸是由带有对应反密码子的内源tRNA编码的天然氨基酸。
B.各密码子的泳道线型分析。
图5(补充性图4)示出由ESI-MS证实的SORT中特异性氨基酸替换
在使用UUU(Lys)且在1mM 3的存在下SORT后分离T4溶菌酶。WT T4溶菌酶预期质量:19512.2Da,实测质量:19510±2.0Da。WT T4溶菌酶Lys→3单突变预期质量:19622.3Da,实测质量:19620±2.0Da。
图6(补充性图5)示出经由SORT-M引入3(0.1mM)对细胞无毒性
用两种质粒来转化化学感受态DH10B细胞:PylRS表达所必需的pBKwtPylRS,以及PyltRNAXXX表达所需的并且在阿拉伯糖控制下表达溶菌酶的pBAD_wtT4L_MbPylTXXX质粒。在37℃下、1ml SOB培养基中恢复细胞一小时,之后等分抽样至10ml LB-KT(具有50μg ml-1卡那霉素和25μg ml-1四环素的LB培养基),并且过夜孵育(37℃,250rpm,12h)。将过夜培养物(OD600≈3)在补充有不同浓度(0mM,0.1mM,0.5mM)3的10mL LB-KT1/2(具有25μg ml-1卡那霉素和12.5μg ml-1四环素的LB培养基)中稀释至OD600约0.3。将这些培养物的200μL等分试样转移入96孔板,并且使用酶标仪Infinite 200Pro(TECAN)来测量OD600。每10min测量各样品的OD600,其中测量之间具有线性1mm振荡。
图7(补充性图6)示出在蛋白质组中经由SORT-M、在3的不同浓度下、响应于AAA密码子来引入3的时间依赖性变化的测量
用两种质粒转化化学感受态DH10B细胞:PylRS表达所必需的pBKwtPylRS,以及PyltRNAUUU表达所需的pBAD_wtT4L_MbPylTUUU质粒。pBAD_wtT4L_MbPylTUUU质粒还含有用于T4溶菌酶表达的基因,该基因在阿拉伯糖诱导型启动子下游。在转化后,在37℃下、1ml SOB培养基中恢复细胞一小时,之后在10ml LB-KT(具有50μg ml-1卡那霉素和25μg ml-1四环素的LB培养基)中接种。将培养物过夜孵育(37℃,250rpm,12h),并且随后在补充有不同浓度(0mM,0.1mM,0.5mM)3的30mL LB-KT1/2(具有25μg ml-1卡那霉素和12.5μg ml-1四环素的LB培养基)中将培养物稀释至OD600约0.3。孵育(37℃,250rpm)培养物1h,此时OD600达到大约0.6。将2ml培养物等分试样收集在针对三个培养物中每个的单独管中。这是前诱导培养物(在凝胶图像中标为1的泳道)。随后,以0.2%(v/v)的终浓度添加阿拉伯糖以诱导T4溶菌酶的表达,并且每小时收集2ml培养物等分试样(标为2、3和4的泳道对应于诱导后1h、2h和3h的培养物收集)。对于每个收集的培养物而言,通过在4℃下离心使细菌细胞沉淀,使用用冰预冷的PBS(3×1ml)漂洗细菌细胞,并且随后将沉淀物在-20℃下冷冻和存储。然后在200μl用冰预冷的PBS中融化沉淀物,并且通过超声处理(9×10s开/20s关、70%功率)来裂解。通过在15,000RPM、4℃下离心30分钟使裂解物澄清。将上清液转移至新的1.5mL管。将50μl上清液转移至新管以用于标记反应,并且将剩余物在液氮中冷冻和存储在-80℃下。将0.5μl 2mM 4a添加至50μl上清液,并且在25℃下孵育裂解物1小时。在1h后,添加补充(6mM BCN和5%BME)的17μl 4×LDS样品缓冲液,并且通过温和涡旋混合。孵育样品10min,之后在90℃下煮沸10min。通过4-12%SDS-PAGE来分析样品,并且使用Typhoon Trio磷光影像分析仪(GE Life Sciences)来获得荧光图像
图8示出在多样的密码子处将3位点特异性引入蛋白质以及在人类细胞中使用SORT-M来特异性标记蛋白质组。(a)Western印迹分析证实在HEK293T细胞中由带有C-端HA标签的琥珀终止密码子隔开的mCherry-EGFP融合蛋白(mCh-TAG-EGFP-HA)的有效、依赖氨基酸的表达。抗FLAG检测加标签的PylRS(b)在HEK293细胞中用4a(在50μL PBS中20μM,1h,室温)特异性标记mCh-TAG-EGFP-HA(免疫沉淀自106个细胞)证实3已引入蛋白质。(c)3的SORT-M标记,使用0.5mM的3、由六种不同PylRS/PyltRNAXXX突变体引导的,3在哺乳动物细胞全蛋白质组上在统计学上引入新合成的蛋白质。用4g标记(在PBS中20μM,1h,室温,如上述)。括号中的氨基酸是由带有对应反密码子的内源tRNA编码的天然氨基酸。
图9(补充性图11)示出
A.来自图8的完整印迹。
B.来自图10的完整印迹。
图10示出将氨基酸3位点特异性引入在黑腹果蝇(Drosophila melanogaster)中产生的蛋白质。(a)由双荧光素酶报告基因证实3的引入。在存在或缺乏10mM 1或10mM 3的情况下,对来自10只表达Triple-Rep-L的雌蝇的卵巢提取物进行双荧光素酶测定。数据示出来自3个生物学重复中1个的代表性实例。误差线表示单个生物学重复中3个技术性重复的标准差。(b)在表达PylRS/PyltRNACUA的果蝇中将3(或1)位点特异性引入GFP_TAG_mCherry-HA。通过抗HA Western印迹来检测来源于非天然氨基酸引入的全长蛋白质。(c)用四嗪探针特异性标记编码的3。用无氨基酸、氨基酸1(500只果蝇)或氨基酸3(100只果蝇)饲喂果蝇。为了产生可比较的报告基因蛋白量,用1饲喂5倍的果蝇。自裂解的卵巢、用抗GFP颗粒来免疫沉淀含非天然氨基酸的全长蛋白。漂洗标记的颗粒(4g,4μM,200μL PBS,室温,2h)。通过抗HA印迹来检测全长蛋白质,并且在荧光扫描仪上成像的相同凝胶显示引入3而非1的蛋白质特异性荧光标记,从而证实引入的氨基酸的身份。
图11(实施例6)在遗传编码的非天然氨基酸1和2处特异性的蛋白标记。(a)在钙调蛋白中遗传编码的1而非2被用探针3特异性标记。考马斯和荧光图像证实标记的特异性,并且在标记前(黑色,预期质量:17875,实测质量:17874)和标记后(红色,预期质量:18553,实测质量:18552)的ESI MS证实反应是定量的。(b)在钙调蛋白中遗传编码的2而非1被用探针4特异性标记。考马斯和荧光图像证实标记的特异性,并且在标记前(黑色,预期质量:17930,实测质量:17930)和标记后(绿色,预期质量:18484,实测质量:18485)的ESI MS证实反应是定量的。未示出原始(解卷积前)的ESI-MS谱。
图12(实施例6)在钙调蛋白的位置1和40处引入1和2以及特异性标记的动力学。(a)在带有ribo-Q1、O-gst-cam1TAG-40AGTA、PylRS/tRNAUACU对和MjPrpRS/tRNACUA对的大肠杆菌中进行表达。分别以4mM和1mM使用氨基酸1和2。(b)CaM11240与3和4反应的标记时程。通过以凝胶荧光和迁移率变动跟踪各反应2h。
图13(实施例6)30分钟内钙调蛋白的协调、定量一锅双标记。(a)CaM11240的染料依赖性标记;在第一标记后,在泳道4中顺序标记、使用纯化,在泳道5中顺序标记、不使用纯化,在泳道6中一锅双标记。(b)一锅蛋白质标记的ESI-MS,在标记前(黑色,预期质量:18000,实测质量:18000),在标记后(金色,预期质量:19233,实测质量:19234)。未示出原始(解卷积前)的ESI-MS谱。
图14示出方案A
图15(补充性图15)示出果蝇GFP-琥珀-mCherry-HA的氨基酸和DNA序列。
GFP(氨基酸残基1-238),琥珀密码子在位置248,mCherry(氨基酸残基255-489),HA标签(氨基酸残基491-499),Myc标签(氨基酸残基500-509),His标签(氨基酸残基510-515)和SV40NLS(氨基酸残基523-528)。
图16示出示例性氨基酸Nε-[((2-甲基环丙-2-烯-1-基)甲氧基)羰基]-l-赖氨酸的结构。
实施例——实施方案的描述
尽管本文已参考附图详细地公开了本发明的说明性实施方案,但要理解本发明不限于精确的实施方案,并且在不脱离由随附权利要求和它们的等价物限定的本发明范围的情况下,可由本领域技术人员实现本文的各种变化和修改。
化学合成——通用方法
所有化学药品和溶剂均购自Sigma-Alrich、Alfa Aesar或Fisher Scientific,并且除非以其他方式规定,否则使用时不用进一步纯化。在涂有硅石(Merck TLC60F-254)的铝片上进行通过薄层色谱(TLC)的定性分析。在短波长紫外灯(254nm)下使斑点显影,或者用碱性含水高锰酸钾、乙醇茚三酮或香草醛使斑点染色。在硅胶60(230-400筛目)上用指定溶剂系统进行快速柱色谱。
在Agilent 1200机器上进行LC-MS分析。使用的溶剂由0.2%于水中的甲酸(缓冲液A)和0.2%于乙腈中的甲酸(缓冲液B)组成。使用Phenomenex Jupiter C18柱(150×2mm,5μm)进行LC,并且使用可变波长监测。记录至最接近0.1min的保留时间(Rt)和至最接近0.01质量单位的m/z比率。对小分子LC梯度使用以下程序:0-1min(A:B 10:90-10:90,0.3mL/min),1-8min(A:B 10:90-90:10,0.3mL/min),8-10min(A:B 90:10-90:10,0.3mL/min),10-12(A:B 90:10-10:90,0.3mL/min)。
LC之后,以ESI模式在6130四极质谱仪上进行质谱分析,并且以阳离子和阴离子模式两者记录。在Bruker 400MHz仪器上进行NMR分析。所有相对于TMS的报告的化学位移(δ)用使用的氘化溶剂中剩余质子来表示:d1-三氯甲烷(1Hδ=7.26ppm,13Cδ=77.16ppm),d6-二甲基亚砜(1Hδ=2.49ppm,13Cδ=39.52ppm),D2O(1Hδ=4.70)。总是进行APT或二维实验(COSY,HSQC)以提供用于需要分析处的额外信息。以Hz形式给出偶联常数,并且如下描述:单峰-s,二重峰-d,三重峰-t,四重峰-q,宽单峰-br,多重峰-m,双二重峰-dd等以及它们的组合。
在大肠杆菌中表达、纯化和用位点特异性引入的3标记蛋白质
自大肠杆菌表达和纯化sfGFP-3使用pBK-MbPylRS和psfGFP150TAG PylT共转化电感受态大肠杆菌DH10B细胞14,26。在37℃下、S.O.B.(1ml,补充有0.2%葡萄糖)中恢复转化的细胞1h,并且转化的细胞用于接种至含50μg/ml卡那霉素和25μg/ml四环素的LB(LB-KT)。在37℃、250r.p.m下过夜振荡孵育细胞。1mL过夜培养物用于接种至100mL LB-KT1/2,然后孵育日间培养物(37℃,250r.p.m)。在O.D.600约0.3时,培养物被等分且用或3(1mM)或H2O(500μL)补充并且进一步孵育(37℃,250r.p.m)。在O.D.600约0.6时,通过添加阿拉伯糖(0.2%)来诱导蛋白质表达,在4h后,通过离心(4000r.p.m,20min)收获细胞,并且将沉淀物冷冻直至再次使用。
在冰上融化冷冻的细菌沉淀物,并且在2.5mL裂解缓冲液(50μg/mL DNAse1,Roche抑制剂鸡尾酒和20mM咪唑)中重悬沉淀物。孵育细胞(4℃,30分钟),然后通过离心(16000g,4℃,30分钟)来使细胞澄清。将澄清的裂解物转移至新管,并且添加100μL Ni-NTA浆液。搅动孵育混合物(4℃,1h),然后通过离心(1000g,4℃,5min)收集混合物。在500μL漂洗缓冲液(10mM Tris-HCL,40mM咪唑,200mM NaCl,pH 8)中使颗粒重悬三次,并且通过离心(1000g,4℃,5min)来收集颗粒。最后,在100μL洗脱缓冲液(10mM Tris-HCL,300mM咪唑,200mM NaCl,pH 8)中使颗粒重悬,通过离心(1000g,4℃,5min)沉淀,并且将上清液收集入新管。用100μL洗脱缓冲液重复洗脱三次。通过4-12%SDS-PAGE和LC-MS来分析纯化的蛋白质。
蛋白质质谱
使用Agilent 1200LC-MS系统,另外用6130四极质谱仪进行ESI-MS。溶剂系统由0.1%于H2O中的甲酸(作为缓冲液A)和0.1%于乙腈(MeCN)中的甲酸(作为缓冲液B)组成。在214nm和280nm监测蛋白质紫外吸光度。以阳离子模式进行蛋白质MS获取,并且通过在MS Chemstation软件(Agilent Technologies)内解卷积来计算总蛋白质量。
体外标记纯化的sfGFP150-3
将4a(10摩尔当量,来自2mM于DMSO中的储液)添加至纯化的sfGFP150-1或sfGFP150-3蛋白(约30μM,在洗脱缓冲液中)。通过抽吸数次使反应物混合,然后在室温下孵育混合物2小时,通过ESI-MS分析样品。孵育后通过4-12%SDS-PAGE分离蛋白质,并且通过使用Typhoon Trio磷光影像分析仪(GE Life Sciences)来分析蛋白。
sfGFP150-3和sfGFP150-NorK标记的时程以及速率常数测定
在室温下通过添加20nmol四嗪染料缀合物4a(10μl 2mM于DMSO中的溶液)来标记2nmol sfGFP-3(10.6μM),通过抽吸数次使样品混合。在不同时间点,从溶液中取8μL等分试样,并且将其用700倍过量二环[6.1.0]壬-4-炔-9-基甲醇(BCN)淬灭且浸没入液氮。用补充有5%β-巯基乙醇的NuPAGE LDS样品缓冲液混合样品,样品被加热10min至90℃并且通过4-12%SDS page分析。通过使用Typhoon Trio磷光影像分析仪(GE Life Sciences)扫描荧光条带来量化标记蛋白质的量。用ImageQuantTM TL软件(GE Life Sciences)使用橡胶带背景扣除来量化条带。通过将数据拟合至单指数方程来测定速率常数。计算观测的速率k'除以4a的浓度得到反应的速率常数k。进行三重测量。使用Kaleidagraph软件(Synergy Software,Reading,UK)执行所有数据处理。为了比较带有Ne-5-降冰片烯-2-基氧羰基-L-赖氨酸(NorK,PylRS的已知底物)的sfGFP的标记速率,使用11.25mM在位置150处带有NorK的sfGFP(SfGFP-NorK)和20当量的4a以相似方式测定速率。
pBAD_wtT4L_MbPylTXXX质粒构建
用NcoI和KpnI限制性内切酶消化pBAD_T4L83TAG_MbPylTCUA。相同的限制性内切酶还用于消化来自(D67)pBAD_wtT4L的野生型T4溶菌酶。使用T4DNA连接酶将插入物和骨架以3:1的比率连接(室温,2小时),转化入化学感受态DH10B细胞并且在四环素琼脂平板上生长(37℃,18小时)。挑取单菌落,并且通过DNA测序(GATC Gmbh.)确认正确序列,此步骤创建pBAD_wtT4L_MbPylTCUA。通过DNA测序确认所有最终构建物。
在表达T4溶菌酶的大肠杆菌中经由SORT在蛋白质组中引入3
用pBAD_wtT4L_MbPylTXXX质粒(2μL,是PyltRNAXXX表达所必需的并且在阿拉伯糖控制下表达T4溶菌酶)和pBKwtPylS质粒(2μL,是PylRS表达所必需的)双转化电感受态大肠杆菌DH10B细胞(50μL),或者用单独的pBAD_wtT4L_MbPylTXXX单转化。在37℃下、1mL S.O.B.(补充有0.2%葡萄糖)中恢复转化的细胞1h。100μL恢复物用于接种5mL LB-KT(50μg/ml卡那霉素和25μg/ml四环素)或LB-T(25μg/ml四环素)。过夜孵育(37℃,250r.p.m.)培养物。1mL各过夜培养物用于接种15mL1/2强度含抗生素的培养基LB-T或LB-KT。在37℃下孵育培养物直至O.D.600达到约0.3,在此时将各培养物分成5mL等分试样并且用3(终浓度0.1mM)或H2O(50μL)补充。然后,孵育(37℃,250r.p.m.)培养物。在O.D.600为0.6时,通过添加阿拉伯糖(终浓度0.2%)引发T4溶菌酶表达,并且孵育培养物又4小时。通过离心(4000rpm,4℃,20分钟)收获细胞,然后在1mL用冰预冷的PBS中重悬细胞三次,并且通过离心(4000rpm,4℃,20分钟)收集细胞。将最终细菌沉淀物立即冷冻存储。
大肠杆菌:使用四嗪染料缀合物化学选择标记加有标签3的蛋白质组
在500μL PBS中重悬冷冻细菌沉淀物,并且使用水浴超声破碎仪裂解(能量输出7.0,90s总超声时间。10s冲击和20s间隙,Misonix超声破碎仪3000)。通过离心(4℃,14000r.p.m.,30min)使裂解物清澈,并且将上清液抽吸至新管。添加4a(2mM,储备于DMSO中,终浓度至20μM)至50μL清澈的细胞裂解物。通过抽吸数次使反应混合,然后在暗处(室温,1h)孵育样品。在此后,添加17μL补充(6mM BCN和5%BME)的4×LDS样品缓冲液,并且通过温和涡旋混合。孵育样品10min,之后在90℃下煮沸10min。通过4-12%SDS-PAGE分析样品,并且使用Typhoon Trio磷光影像分析仪(GE Life Sciences)来获得荧光图像。
用于荧光标记大肠杆菌蛋白质的相同规程应用于所有四嗪染料缀合物。
在HEK293细胞中位点特异性引入3以及使用四嗪探针的化学选择标记
在HEK细胞中位点特异性引入3
将HEK293细胞(ATCCCRL-1573)铺板于24孔板上,并且使其生长至接近汇合。用pMmPylS-mCherry-TAG-EGFP-HA构建物和p4CMVE-U6-PylT构建物使用Lipofectamine 2000(Invitrogen)转染细胞18。在用或不用1mM 3或用1mM 1生长16hr后,在冰上使用RIPA缓冲液(Sigma)裂解细胞。将裂解物旋离下来,并且将上清液添加至4×LDS样品缓冲液(Life technologies)。通过SDS-PAGE分离出样品,将样品转移至硝酸纤维素膜并且使用第一大鼠抗HA(克隆3F10,Roche1867423号)和小鼠抗FLAG(克隆G191,Abnova,cat.MAB8183)进行印迹,第二抗体是抗大鼠(Invitrogen,A11077)和抗小鼠(Cell Signaling Technologies,7076S号)。
从HEK 293细胞中标记位点特异性引入的3
使用TransIT-293转染试剂、根据制造商的规程用7.5μg p4CMVE-U6-PylT和7.5μg pPylRS-mCherry-TAG-EFGP-HA转染贴附的HEK293T细胞(ATCCCRL-11268;每次免疫沉淀4×106个)18,并且在DMEM/10%FBS中培养细胞48小时,在指示处用0.5mM 1或2mM 3补充。用PBS漂洗细胞两次,并且在冰上于1mL裂解缓冲液(150mM NaCl,1%Triton X-100,50mM Tris HCl(pH 8.0)中裂解细胞。在通过离心(在16000g下10min)使裂解物澄清后,每次转染使用50μLμMACS HA标签微珠(Miltenyl Biotec)来捕获加HA标签的蛋白质,用0.5mL RIPA(150mM NaCl,1%Igepal CA-630,0.5%脱氧胆酸钠,0.1%SDS,50mM Tris HCl(pH 8.0)和0.5mL PBS(pH 7.4)漂洗蛋白。用50μl PBS(pH 7.4)和20μM4a孵育微珠悬浮液1小时,并且随后用0.5mL RIPA漂洗以去除过量染料。将加HA标签的蛋白质使用SDS样品缓冲液从微珠上洗脱并且在4-12%Bis-Tris PAGE凝胶(Invitrogen)上分离,使用Typhoon成像仪(GE Healthcare)成像,并且随后用DirectBlue染色或转移以用抗HA标签pAb-HRP-DirecT(MBL)进行western印迹。
从哺乳动物细胞中表达和纯化SfGFP
在10cm组织培养皿中使用PEI用15μg DNA转染HEK293T,并且用3(0.5μM)孵育72小时。将细胞用PBS漂洗两次,并且在1mL RIPA缓冲液中裂解。将清澈的裂解物添加至50μLGFP-M(ChromoTek)并且孵育4h。将微珠用1mL RIPA、1mL PBS、1mL PBS+500mM NaCl、1mL ddH2O漂洗,并且在1%乙酸/ddH2O中洗脱。将纯化的蛋白质用2μM 4a标记,并且上样至4-12%Bis-Tris PAGE凝胶上。在Typhoon成像仪上检测标记4a的sfGFP的荧光,并且随后将凝胶用DirectBlue染色。
果蝇质粒,转基因果蝇以及培养
对于在此研究内的所有果蝇实验未使用随机化或盲法。
转基因果蝇系代的质粒构建
使用QuikChange诱变试剂盒并且用pSG108(pJet 1.2-U6-PylT,馈赠自S.Greiss)作为模板来突变PyltRNACUA反密码子。此PyltRNACUA反密码子含有融合至果蝇U6-b启动子的PylT基因,该PylT没有其3'端CCA。引物FMT19和FMT20用于产生PyltRNATGC以编码丙氨酸密码子(创建pFT18);引物FMT23和FMT24用于产生PyltRNAGCT以编码丝氨酸密码子(创建pFT20);引物FMT27和FMT28用于产生PyltRNACAG以编码亮氨酸密码子(创建pFT22),并且引物FMT29和FMT30用于产生PyltRNACAT以编码甲硫氨酸密码子(创建pFT23)。将突变的tRNA表达框使用EcoRI和HinDIII从pFT18、pFT20、pFT22和pFT23亚克隆入pUC18,然后使用AsiSI、BamHI和BglII多聚化以创建2拷贝然后是4拷贝tRNA。使用AsiSI和MluI将4拷贝形式的tRNA框亚克隆入pSG118,以创建pFT58(Ala)、pFT60(Ser)、pFT62(Leu)和pFT63(Met)。pSG118含有马氏甲烷八叠球菌(M.mazei)PylRS基因20。
果蝇系和培养条件
使用果蝇胚胎注射服务(BestGene公司)由P元件插入来创建转基因系。使用下述质粒产生系:pFT58(Ala)、pFT60(Ser)、pFT62(Leu)和pFT63(Met)。nos-Gal4-VP16(Bloomington 4937)和MS1096-Gal4(Bloomington 8860)用作Gal4驱动子(driver)。所有果蝇在25℃下、在标准Iberian介质上生长。用非天然氨基酸饲喂果蝇,通过使干酵母与于dH2O中稀释适当浓度(通常10mM)的氨基酸混合以制造浆糊来饲喂。自雌蝇制备卵巢,所述雌蝇在用有或无氨基酸的酵母糊补充的Iberian果蝇食物上生长最少48小时。对于蛋白质组标记实验而言,具有构建物FT58、FT60、FT62和FT63的转基因雄果蝇分别与nos-vp16-GAL4处女蝇交配以产生FT58/nos-vp16-GAL4、FT60/nos-vp16-GAL4、FT62/nos-vp16-GAL4和FT63/nos-vp16-GAL4。
在黑腹果蝇(D.melanogaster)中位点特异性引入3
荧光素酶测定
卵巢来自用3、1或无氨基酸饲喂的重组有nos-Gal4-VP16的Triple Rep-L果蝇中的10只雌蝇,将卵巢在100μl 1×被动裂解缓冲液中解剖并且将其处理以用于先前描述的荧光素酶测定20。
免疫沉淀以及标记位点特异性引入的3
将来自100只雌蝇(用于对照和3)或500只雌蝇(用于1)的卵巢在PBS中解剖,然后将其在含1×完全蛋白酶抑制剂鸡尾酒(Roche)的300或1500μl RIPA缓冲液中裂解。将样品取入4×LDS缓冲液作为总裂解物对照,然后剩余物用于按照制造商的说明书用GFP-TRAP琼脂糖珠(Chromotek)进行免疫沉淀。免疫沉淀(IP)的总体积是3ml。在过夜孵育后,将微珠用RIPA缓冲液漂洗2次,然后用PBS漂洗2次。对于四嗪标记而言,将微珠重悬于200μl PBS+4μM 4g中,并且在室温下在滚动器上孵育2小时。将微珠用500μL漂洗缓冲液漂洗3次,然后重悬于4×LDS样品缓冲液中。
实施例1——Nε-[((2-甲基环丙-2-烯-1-基)甲氧基)羰基]-L-赖氨酸3的合成
一类在蛋白质标记中有用的反应是非常快速和特异的在应变烯烃(或炔烃)与四嗪之间的逆电子需求狄尔斯-阿尔德反应21-25。
尽管我们和其他人先前已经由遗传密码扩展来编码带有应变烯烃、炔烃和四嗪的非天然氨基酸,并且证实了它们经由逆电子需求狄尔斯-阿尔德反应用于位点特异性蛋白质标记的用途26-30,但至今使用的所有分子都相当大。我们先前已示出各种赖氨酸的氨基甲酸酯衍生物是PylRS的良好底物31,并且已证实不同于3,3二取代环丙烯32,24,1,3二取代环丙烯能与四嗪有效反应22。因此,我们设计并合成了一种带有1,3二取代环丙烯的赖氨酸氨基甲酸酯衍生物(Nε-[((2-甲基环丙-2-烯-1-基)甲氧基)羰基]-L-赖氨酸3,图1b),用于引入蛋白质并且用四嗪标记。
甲基环丙-2-烯-1-基}甲氧基)羰基]-L-赖氨酸(3)的合成
方案1.Nε-[({2-甲基环丙-2-烯-1-基}甲氧基)羰基]-L-赖氨酸(3)的合成。试剂和条件:i.Rh2(OAc)4,丙炔,CH2Cl2,4℃至室温,产率75%;ii.DIBAL-H,CH2Cl2,0℃至室温;iii.4-对硝基苯基氯甲酸酯,N,N-二异丙基乙胺(Hünig's碱),CH2Cl2,室温,产率73%;iv.Fmoc-Lys-OH,N,N-二异丙基乙胺,THF/DMF,4℃至室温,产率82%;v.NaOH,THF/H2O,室温,产率68%。
i.乙基2-甲基环丙-2-烯-1-羧酸酯S1
将100mL 2颈圆底烧瓶用CH2Cl2(2mL)和乙酸铑(442mg,1mmol,0.05当量)装载,并且配有干冰冷凝器。将丙炔(大约10mL)冷凝入乙酸铑悬浮液,并且使烧瓶降入到水浴(20℃)里,得到丙炔的稳定回流。使用注射泵将重氮乙酸乙酯(2.1mL,20mmol,1当量)在1h内逐滴添加至搅拌的丙炔溶液。在室温下搅拌反应又10分钟,通过TLC分析显示在此后反应完成。然后,通过硅胶快速柱色谱用戊烷和二乙醚(90:10)洗脱来纯化环丙烯产物。这给出期望产物的无色挥发性液体S1(1.9g,产率75%)。1H NMR分析δH(400MHz,CDCl3)6.35(1H,t,J 1.4),4.18-4.09(2H,m),2.16(3H,d,J 1.3),2.12(1H,d,J 1.6),1.26(3H,t,J 7.1);LRMS m/z(ES+)127.2[M+H]+。
这些值与文献的一致性良好。{Liao,2004#1}
ii.和iii.(2-甲基环丙-2-烯-1-基)甲基(4-硝基苯基)碳酸酯S3
在-10℃下,将DIBAL-H(22.5mL 1M于CH2Cl2中的溶液,22.5mmol,1.5当量)逐滴添加至于CH2Cl2(15mL)中的环丙烯酯S1(1.9g,15mmol,1当量)搅拌溶液。在-10℃下搅拌反应20分钟,之后用谨慎添加H2O(1mL)、然后添加NaOH(1mL 1M于H2O中的溶液)和H2O(2.3mL)来淬灭。在室温下搅拌混合物又2h,之后将混合物干燥(Na2SO4)和过滤。将N,N-二异丙基乙胺(3.9mL,22.5mmol,1.5当量)添加至滤出液(含粗制环丙烯醇S2),接着添加4-对硝基苯基氯甲酸酯(3.3g,16.5mmol,1.1当量)。在室温下搅拌18小时后形成显著无色沉淀物,并且TLC分析显示了粗制环丙烯醇S2的完全消耗。用CH2Cl2稀释反应,然后上样至硅胶上干燥,借此通过硅胶柱色谱用乙酸乙酯和己烷(20:80)洗脱来纯化活化的碳酸酯S3。这给出期望的环丙烯碳酸酯的无色油状物S3(2.7g,经2个步骤产率73%)。1H NMR分析δH(400MHz,CDCl3)8.28(2H,d,J 9.2),7.39(2H,d,J 9.2),6.62(1H,s),4.21(1H,dd,J 10.9,5.3),4.14(1H,dd,J 10.9,5.3),2.18(3H,d,J 1.3),1.78(1H,td,J 5.3,1.3)。
iv.Nα-(Fmoc)-Nε-(((2-甲基环丙-2-烯-1-基)甲氧基)羰基)-L-赖氨酸S4
将Fmoc-Lys-OH-HCl(6.7g,16.5mmol,1.5当量)溶解于THF(30mL)和DMF(10mL)中,将Hünig’s碱(9.0mL,55.0mmol,5当量)添加至此溶液,接着添加环丙烯碳酸酯S3(2.7g,11.0mmol,1当量),在添加碳酸酯时立即观察到黄色颜变。在室温下搅拌反应6小时,在此后通过TLC分析示出的起始物质消耗来判定反应完成。将粗制反应混合物上样至硅胶上干燥,并且通过硅胶柱色谱用乙酸乙酯、己烷和乙酸(50:49:1,然后99:0:1)洗脱来纯化主要产物。这给出期望产物的无色胶状物S4(4.3g,产率82%)。1H NMR分析δH(400MHz,CDCl3)7.77(2H,t,J 7.6),7.65-7.55(2H,m),7.39(2H,t,J 7.6),7.31(2H,t,J 7.3),6.54(1H,s),5.68-5.57(1H,m),4.84(1H,br-s),4.44-4.32(2H,m),4.22(1H,t,J 7.0),3.98-3.87(1H,m),3.17-3.09(2H,m),2.15-2.06(6H,m),1.99-1.86(1H,m),1.84-1.70(1H,m),1.68-1.59(1H,m),1.58-1.34(2H,m);LRMS m/z(ES+)479.3[M+H]+,501.3[M+Na]+,m/z(ES-)477.2[M-H]-。
v.Nε-[({2-甲基环丙-2-烯-1-基}甲氧基)羰基]-L-赖氨酸 3
将Nα-(Fmoc)-Nε-(((2-甲基环丙-2-烯-1-基)甲氧基)羰基)-L-赖氨酸S4(3.5g,7.0mmol,1当量)溶解于THF和H2O(3:1 40mL)中,将氢氧化钠(0.9g,22.6mmol,3.1当量)添加至此溶液。在室温下搅拌反应8小时,在此后通过LC-MS分析判定反应完成。用H2O(100mL)稀释反应混合物,并且通过添加HCl(1M)将pH调节至约5。用Et2O(5×100mL)漂洗含水溶液,然后将溶液浓缩至干燥,产生无色固体。通过制备HPLC来纯化固体,产物部分被合并,并且通过冷冻干燥去除溶剂。这给出无色固体形式的Nε-[({2-甲基环丙-2-烯-1-基}甲氧基)羰基]-L-赖氨酸3。δH(400MHz,D2O)6.45(1H,s),3.90-3.61(2H,m),3.09(1H,t,J 6.4),2.98-2.86(2H,m),1.92(3H,s),1.52-1.37(2H,m),1.37-1.22(2H,m),1.21-1.08(2H,m),0.83(1H,d,J 5.2)。LRMS m/z(ES+)257.2[M+H]+,m/z(ES-)255.2[M-H]-。δC(100MHz,D2O)101.1(CH),72.3(CH2),55.9(CH),40.2(CH2),34.3(CH2),28.9(CH2),20.3(CH2),16.6(CH3),10.8(CH)HRMS(ES+)实验值:(M+Na)+279.1302。C12H20O4N2Na所需M+,279.1315。
实施例2——在大肠杆菌中编码3的位点特异性引入
我们证实了使用PylRS/tRNACUA对和在位置150处带有琥珀密码子的SfGFP基因将3响应于琥珀密码子有效地和位点特异性引入入重组蛋白(补充性图2a)。每升培养物中蛋白质的产量是8mg,这大于用已良好确立的PylRS有效底物Nε-[(叔-丁氧基)羰基]-L-赖氨酸1得到的量(每升培养物4mg)33。在位置150处带有3的SfGFP(SfGFP-3)的电喷雾离子化质谱证实非天然氨基酸的引入(补充性图2b)。SfGFP-3被用荧光四嗪探针4a特异性标记,而SfGFP-1未被标记(补充性图2b)。通过荧光成像和质谱两者判断在30分钟内2nmol SfGFP-3被用10当量4a定量标记(补充性图2b)。用4a标记SfGFP-3的二级速率常数是27±1.8M-1s-1(补充性图2c)26。
因为PylRS不识别其同族tRNA的反密码子34,所以可能改变此tRNA的反密码子以编码不同的密码子。我们创建了一种新的tRNA,其中将PyltRNACUA的反密码子从CUA变换为UUU(补充性表1),以编码一组赖氨酸密码子。我们将0.1mM 3添加至含PylRS、PyltRNAUUU和T4溶菌酶基因的细胞。在T4溶菌酶表达后,我们用四嗪探针4a(20mM)检测了裂解物中带有3的蛋白质(1h,补充性图3)。对照实验显示可观测的标记需要合成酶和tRNA的存在,并且电喷雾离子化质谱证实3引入在T4溶菌酶中的赖氨酸位置中(补充性图4)。3(0.1mM或0.5mM)的添加对细胞生长有很小效应或无效应(补充性图5),这表明该氨基酸在使用的浓度下无毒性,并且在添加氨基酸的1h内存在大量标记(补充性图6)。
实施例3——在人类细胞中3的遗传编码
在携带PylRS/tRNACUA对和mCherry-TAG-EGFP-HA(mCherry基因与EGFP基因之间的融合,具有C-端HA标签,由琥珀终止密码子(TAG)隔开)的HEK293细胞中表达全长mCherry-3-GFP-HA18。仅在3的存在下检测到全长蛋白(图8a.完整凝胶在补充性图11中)。mCherry-3-EGFP-HA被用4a选择性标记,而mCherry-1-EGFP-HA未被标记(图8b)18,这证实了在人类细胞中用PylRS/tRNACUA对3的位点特异性引入。
实施例4——在黑腹果蝇中3的遗传编码
我们证实了在黑腹果蝇中可将3位点特异性引入蛋白质。为了实现这点,我们使用含有PylRS/tRNACUA对(其中tRNA利用U6启动子遍在表达,并且使用nos-vp16-GAL4驱动子将UAS-PylRS引导至卵巢表达)和双荧光素酶报告基因(在萤火虫荧光素酶与海肾荧光素酶之间带有琥珀密码子)的果蝇20。我们观察到依赖1或3添加的强荧光素酶信号,并且使用3的双荧光素酶信号更强。这些实验证实3被果蝇吸收,并且与已知PylRS的极佳底物1相比,在体内3响应于琥珀密码子被更有效引入(图10a)。可通过饲喂补充有10mM氨基酸3的食物来供应3。在额外实验中,我们通过western印迹证实3被有效引入在卵巢中表达(图10b)的GFP-TAG-mCherry-HA构建物(补充性图15)20,以及用4g特异性荧光标记该引入的氨基酸(图10c)。
实施例5——四嗪-BODIPY FL 4d的合成
方案3.四嗪-生物素4d的合成。试剂和条件:i.HCl二噁烷,室温,产率100%;ii.Bodipy-FL-NHS酯,N,N-二异丙基乙胺,DMF,室温。
i.S6
使用之前报道的程序来合成Boc保护的四嗪S66。将4M于二噁烷(500μL,2.0mmol)中的HCl添加至四嗪S5(8mg,0.02mmol)于DCM(500μL)中的搅拌溶液。在室温下进行反应2h,并且随后在减压下去除溶剂,以产生粉红色固体形式的盐酸伯胺S6(6mg,0.02mmol,100%)。该化合物可不进行任何进一步纯化直接在下一个步骤中使用。
ii.4d
将BODIPY FL琥珀酰亚胺酯(5mg,0.013mmol,Life technologies)和Hünig’s碱(50μl,2.8mmol)添加至四嗪-胺S2(6mg,0.02mmol)于干燥DMF(1mL)中的溶液。在室温下搅拌反应混合物16h。用4ml水稀释反应混合物,通过半制备反向HPLC使用10%至90%的于缓冲液A中的缓冲液B梯度(缓冲液A:H2O;缓冲液B:乙腈)来纯化产物。通过LC-MS确认四嗪-BODIPY FL缀合物4d的真伪和纯度。ESI-MS:[M-H]-,计算值581.38,实验值581.2。
实施例1至5的总结
我们已表征含环丙烯氨基酸3的合成和遗传编码的位点特异性引入,并且证实了在大肠杆菌、哺乳动物细胞和黑腹果蝇中表达的蛋白质中用四嗪探针定量标记3,藉此显示本发明的广泛效用和工业应用。
实施例1至5的补充性参考
1.Gautier,A.等人.Genetically Encoded Photocontrol of Protein Localization in Mammalian Cells.Journal of the American Chemical Society132,4086-4088(2010).
2.Karp,N.A.,Kreil,D.P.&Lilley,K.S.Determining a significant change in protein expression with DeCyder during a pair-wise comparison using two-dimensional difference gel electrophoresis.Proteomics4,1421-1432(2004).
3.Karp,N.A.&Lilley,K.S.Design and analysis issues in quantitative proteomics studies.Proteomics7Suppl 1,42-50(2007).
4.Lilley,K.S.in Current Protocols in Protein Science(John Wiley&Sons,Inc.,2001).
5.Von Stetina,J.R.,Lafever,K.S.,Rubin,M.&Drummond-Barbosa,D.A Genetic Screen for Dominant Enhancers of the Cell-Cycle Regulator alpha-Endosulfine Identifies Matrimony as a Strong Functional Interactor in Drosophila.G3(Bethesda)1,607-613(2011).
6.Lang,K.等人.Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction.Nat Chem4,298-304(2012).
实施例6——蛋白质双标记
将两种不同的分子附接至蛋白质中程序化位点的能力将有利于各种应用(包括FRET)1,2以研究蛋白质结构、构象和动力学。已报道若干用于双重标记蛋白质的方法。一种方法依赖于被特异性标记的一种非天然氨基酸的装配,并组合半胱氨酸巯基标记,但此方法一般受限于不含游离巯基的蛋白质3,4。可将化学连接方法与用于蛋白质标记的单个非天然氨基酸的遗传编码结合5,但这可能限制可被标记的尺寸和/或位点。大概可最一般适用于蛋白质双标记的方法是基于响应于在目标基因中用户定义位点引入的两种不同的密码子来遗传引入两种不同的氨基酸。
用于双标记的理想策略需要:i)将两种不同的非天然氨基酸有效地、在细胞水平上(cellular)引入可在相互正交反应中被标记的蛋白质,以及ii)研制允许将两种分子同时添加至蛋白质的相互正交反应,用于在生理pH、温度和压力下在水介质中快速、定量标记蛋白质。
方案A(图14)示出在生理pH和温度下在水介质中蛋白质的协调、快速、一锅定量双标记。(a)此实例中使用的非天然氨基酸和荧光团。(b)经由相互正交环加成在编码的末端炔烃和编码的环丙烯处的协调标记。
将两种不同的非天然氨基酸在细胞水平上遗传引导地引入蛋白质已被证实响应于琥珀和四联体密码子6、两种不同的终止密码子7,8或两种不同的四联体密码子9。我们先前证实了正交核糖体(ribo-Q1)的开发,该正交核糖体分别使用同族扩大的反密码子tRNA或琥珀抑制子来有效读取四联体密码子和琥珀密码子6。我们证实了吡咯酰-tRNA合成酶/tRNA对和合成开发的MjTyrRS/tRNA对衍生物在它们的氨酰化特异性上是相互正交的,并且它们可用于引导多对非天然氨基酸响应于琥珀和四联体密码子的引入6。最近我们描述了若干在此系统中的主要进展,包括基于吡咯酰-tRNA合成酶(PylRS)/tRNA对的一系列四联体解码tRNA的开发,该PylRS/tRNA对使用开发的正交翻译机制来有效引导非天然氨基酸响应于四联体密码子的引入9。我们证实了使用开发的PylRS/tRNAUACU对和MjTyrRS/tRNACUA对的衍生物、用带有TAG和AGTA密码子以及ribo-Q1的正交信息体来非常有效地引入多对非天然氨基酸的矩阵9。
针对含多对非天然氨基酸的蛋白质的双标记,已研究了有限范围的化学药品。已报道了含叠氮化物和含炔烃氨基酸的引入,以及用基于炔烃和叠氮化物的荧光团来非定量标记它们7,但这对蛋白质双标记不理想;如果编码的叠氮化物和炔烃二者接近,则它们可能在蛋白质中反应形成三唑,这种策略允许遗传引导的蛋白质装订6,但阻止使用探针的标记。此外,由于带有叠氮化物与带有炔烃的探针彼此之间的反应,有效的一锅反应是不可行的。已报道了含酮和含叠氮化物氨基酸的引入8,10,这允许编码的酮与阿尔法效应亲核试剂以及叠氮化物与炔烃探针的一锅反应10。然而,此方法是有问题的,因为当在大肠杆菌中表达时,在许多蛋白质中编码的叠氮化物遭到还原(reduction)8,11,这将阻止定量标记。此外,用阿尔法效应亲核试剂标记酮是非常缓慢的(速率常数大约10-4M-1s-1)并且反应最佳在pH4-5.5下进行12,这限制了它对许多在酸性条件下长时间保存时会变性或沉淀的蛋白质的效用。最近,我们使用我们优化的正交翻译系统将含钝化四嗪的氨基酸13和含降冰片烯的氨基酸14-16遗传装配入蛋白质9。因为钝化四嗪与降冰片烯之间的逆电子需求狄尔斯阿尔德反应的速率非常缓慢,而四嗪能与基于双环壬炔的探针反应且降冰片烯可与活化的四嗪探针反应,所以我们能够使用此方法来特异性和定量地双标记蛋白质9。尽管此方法具有在生理pH、温度和压力下在水介质中进行的优点;但它确实需要顺序的标记步骤(以避免探针之间的逆电子需求反应),这些步骤中的每个都花费若干小时,且步骤之间需要纯化。所有至今报道的用于在遗传编码的非天然氨基酸处双重标记蛋白质的方法都花费数十小时至数十天以达到完成。
双标记蛋白质的理想方法将允许遗传装配的生物正交官能团的快速一锅标记,在生理pH、温度和压力下在水介质中快速进行,并且仅仅通过将标记试剂添加至带有位点特异性引入的生物正交基团的重组蛋白来实施。在含水条件、生理pH下用于一锅标记的一对有希望的相互正交反应是Cu(I)催化的叠氮化物与末端炔烃之间的3+2环加成17,以及应变烯烃与四嗪的逆电子需求狄尔斯阿尔德反应18-23(图11)。应变炔烃和叠氮化物的反应还可与应变烯烃四嗪反应正交,但因为四嗪与应变炔烃反应,所以此方法需要小心调整各反应的速率常数24。目前未证实用于蛋白质标记的3+2环加成与逆电子需求狄尔斯阿尔德反应的组合。
在实施例1至5中我们证实了可使用PylRS/tRNACUA对将含1,3二取代环丙烯的氨基酸2(在实施例1至5中以及在此文件中的别处称作3)有效和位点特异性引入蛋白质25。这种氨基酸不同于光点击反应引入的3,3二取代环丙烯26,其以蛋白质上27M-1s-1的速率常数25与四嗪反应19,27。此处,我们证实将含末端炔烃的氨基酸1和含环丙烯的氨基酸2有效遗传编码入单个蛋白质,以及它们用叠氮化物和四嗪探针的快速、定量、一锅标记(图11)。此工作提供首个在含水条件、生理pH下以一锅法协调双标记蛋白质的方法,并且提供在双标记速度上的阶跃变化,从先前的工作中的数天变化至此处报告的方法中的30分钟。
使含1或2的蛋白质过表达以检验所提出的标记反应的正交性特异性。在钙调蛋白位置1处具有氨基酸1的谷胱甘肽-S-转移酶与钙调蛋白的融合蛋白(GST-CaM)自在1(4mM)存在下生长的细胞表达,所述细胞含有ribo-Q1(开发的正交核糖体6,28,29)、O-gst-cam1TAG(在正交信息体上的谷胱甘肽-S-转移酶(gst)与钙调蛋白(cam)之间的融合基因30,其中用TAG密码子替换cam的首个密码子)和MjPrpRS/tRNACUA(针对响应于TAG密码子引入1研制的合成酶/tRNA对)31。随后通过使用凝血酶在GST与CaM之间的工程化凝血酶切割位点切割来去除GST标签。在Cu(I)催化的点击反应中,用含叠氮化物的荧光团3(2nmol)标记CaM11(在位置1含有1的CaM,约100皮摩)。通过SDS-PAGE由荧光标记蛋白质的定量位移和电喷雾离子化质谱(ESI-MS)两者判断反应是定量的(图11a)。
将含环丙烯氨基酸2位点特异性引入在钙调蛋白的位置40处。在带有PylRS/tRNACUA(其有效引导2的位点特异性引入)25、ribo-Q1和O-gst-cam40TAG,且在2(1mM)的存在下生长的细胞中表达修饰的蛋白质。用含四嗪的荧光团4(2纳摩)来标记CaM240(约100皮摩)(在凝血酶切割GST标签后得到)。通过SDS-PAGE由荧光标记蛋白质的定量位移和电喷雾离子化质谱(ESI-MS)两者判断反应是定量的(图11b)。在导致CaM11被用3定量标记的条件下,CaM240不被3标记(图11a)。类似地,在CaM240被4定量标记的条件下,CaM11不被4标记。这些实验证实在蛋白质中,两种标记试剂与它们的靶标氨基酸定量反应,但不与它们的非靶标非天然氨基酸反应。
接下来我们研究了在相同蛋白质内标记1和2。我们在钙调蛋白的位置1和40位点特异性引入1和2以产生CaM11240(图12)。我们用MjPrpRS/tRNACUA对引导氨基酸1的引入,并且用开发的PylRS/tRNAUACU对引导氨基酸2的引入,这在使用ribo-Q1的正交信息体上有效地解码四联体AGTA密码子。在正交信息体(O-gst-cam1TAG-40AGTA)上的GST-钙调蛋白基因内,在钙调蛋白中位置1和40处响应于UAG和AGTA密码子来引入非天然氨基酸。全长GST-CaM11240的表达取决于添加至大肠杆菌的氨基酸1和2,并且ESI-MS证实氨基酸1和2的遗传引导的引入(图12c)。全长GST-CaM11240的产量为每L培养物约2mg。
为了测定用叠氮化物3或四嗪4定量标记CaM11240所需的时间,我们用2纳摩3或4孵育100皮摩CaM11240,并且通过SDS-PAGE上的迁移率变动和在标记后的荧光成像两者来跟踪各反应(图12b)。这些实验证实荧光团标记在30分钟内完成。
接下来,我们研究了用3和4两者标记CaM11240(图13)。我们首先测试了将4(2纳摩)添加至CaM11240(100皮摩),接着纯化以去除游离的4,并且随后用3(2纳摩)标记(图13a泳道4)。这导致如通过SDS-PAGE迁移率变动和荧光成像判断的有效双标记。接下来,我们通过用4孵育CaM11240 30分钟,然后添加3和点击试剂并且孵育又30min来进行无纯化的顺序标记(图13a泳道5)。这也导致如通过SDS-PAGE迁移率变动和荧光成像判断的有效双标记。最后,我们将4(2纳摩)、3(2纳摩)和点击试剂同时添加至CaM11240(100皮摩)并且孵育30分钟(图13a泳道6)。这再次导致如通过SDS-PAGE迁移率变动和荧光成像判断的有效双标记。在所有双重标记的蛋白质中,我们观察到在以488nm激发时相对于单标记对照,BODIPY-FL荧光减少(比较图13a中的泳道4、5和6与泳道3),这与在凝胶中BODIPY-FL与BODIPY-TMR-X之间的荧光共振能量转移(FRET,resonance energy transfer)一致。ESI-MS进一步证实此协调的一锅规程导致遗传引导的、有效、快速和定量的蛋白质双标记。
总之,在此实施例中,我们示出一种用于表达带有位点特异性引入的炔烃和位点特异性引入的环丙烯的重组蛋白的有效和快速规程。我们证实编码的1,3二取代环丙烯和四嗪探针的逆电子需求狄尔斯阿尔德反应与编码的炔烃和叠氮化物探针的3+2环加成反应彼此相互正交,并且与蛋白质中的官能团相互正交。通过在单个蛋白质中结合炔烃和环丙烯的遗传编码,并且用相互正交反应来标记,我们证实了在生理pH和温度下、在水介质中蛋白质的协调、一锅快速双标记。此策略具有用于对各种研究和应用的双重标记蛋白质的效用,并且可能被扩大至在多样的细胞和生物中对多样的分子进行双标记。
关于实施例6的注解:在实施例6中和在实施例6中论述的对应图(附图)中的化学名称是独立的,并且仅应用于实施例6。除实施例6以外,在此文件剩余部分中的化学名称论述是一致的。例如,有经验的读者将立即理解实施例6的化合物2对应于此文件剩余部分中的化合物3(即,本发明的示例性环丙烯氨基酸)。实施例6的化合物3和4是四嗪化合物。
实施例6的参考
(1)Zhang,J.;Campbell,R.E.;Ting,A.Y.;Tsien,R.Y.Nature Reviews Molecular Cell Biology2002,3,906.
(2)Kajihara,D.;Abe,R.;Iijima,I.;Komiyama,C.;Sisido,M.;Hohsaka,T.Nat Methods2006,3,923.
(3)Brustad,E.M.;Lemke,E.A.;Schultz,P.G.;Deniz,A.A.J Am Chem Soc2008,130,17664.
(4)Nguyen,D.P.;Elliott,T.;Holt,M.;Muir,T.W.;Chin,J.W.J Am Chem Soc2011,133,11418.
(5)Wissner,R.F.;Batjargal,S.;Fadzen,C.M.;Petersson,E.J.J Am Chem Soc2013,135,6529.
(6)Neumann,H.;Wang,K.;Davis,L.;Garcia-Alai,M.;Chin,J.W.Nature2010,464,441.
(7)Wan,W.;Huang,Y.;Wang,Z.;Russell,W.K.;Pai,P.J.;Russell,D.H.;Liu,W.R.Angew Chem Int Ed Engl2010,49,3211.
(8)Chatterjee,A.;Sun,S.B.;Furman,J.L.;Xiao,H.;Schultz,P.G.Biochemistry2013.
(9)Wang,K.;Sachdeva,A.;Cox,D.J.;Wilf,N.W.;Wallace,S.;Mehl,R.A.;Chin,J.W.submitted.
(10)Wu,B.;Wang,Z.;Huang,Y.;Liu,W.R.Chembiochem:a European journal of chemical biology2012,13,1405.
(11)Sasmal,P.K.;Carregal-Romero,S.;Han,A.A.;Streu,C.N.;Lin,Z.;Namikawa,K.;Elliott,S.L.;Koster,R.W.;Parak,W.J.;Meggers,E.ChemBioChem2012,13,1116.
(12)Rotenberg,S.A.;Calogeropoulou,T.;Jaworski,J.S.;Weinstein,I.B.;Rideout,D.Proceedings of the National Academy of Sciences of the United States of America1991,88,2490.
(13)Seitchik,J.L.;Peeler,J.C.;Taylor,M.T.;Blackman,M.L.;Rhoads,T.W.;Cooley,R.B.;Refakis,C.;Fox,J.M.;Mehl,R.A.J Am Chem Soc2012,134,2898.
(14)Lang,K.;Davis,L.;Torres-Kolbus,J.;Chou,C.;Deiters,A.;Chin,J.W.Nat Chem2012,4,298.
(15)Plass,T.;Milles,S.;Koehler,C.;Szymański,J.;Mueller,R.;Wieβler,M.;Schultz,C.;Lemke,E.A.Angewandte Chemie International Edition2012,51,4166.
(16)Kaya,E.;Vrabel,M.;Deiml,C.;Prill,S.;Fluxa,V.S.;Carell,T.Angewandte Chemie International Edition2012,51,4466.
(17)Wang,Q.;Chan,T.R.;Hilgraf,R.;Fokin,V.V.;Sharpless,K.B.;Finn,M.G.J Am Chem Soc2003,125,3192.
(18)Devaraj,N.K.;Weissleder,R.Accounts of Chemical Research2011,44,816.
(19)Yang,J.;J.;Cole,C.M.;Devaraj,N.K.Angewandte Chemie International Edition2012,51,7476.
(20)Blackman,M.L.;Royzen,M.;Fox,J.M.J Am Chem Soc2008,130,13518.
(21)Lang,K.;Davis,L.;Wallace,S.;Mahesh,M.;Cox,D.J.;Blackman,M.L.;Fox,J.M.;Chin,J.W.J Am Chem Soc2012,134,10317.
(22)Borrmann,A.;Milles,S.;Plass,T.;Dommerholt,J.;Verkade,J.M.M.;Wieβler,M.;Schultz,C.;van Hest,J.C.M.;van Delft,F.L.;Lemke,E.A.ChemBioChem2012,13,2094.
(23)Schoch,J.;Staudt,M.;Samanta,A.;Wiessler,M.;Jaschke,A.Bioconjug Chem2012,23,1382.
(24)Karver,M.R.;Weissleder,R.;Hilderbrand,S.A.Angew Chem Int Ed Engl2012,51,920.
(25)Bianco,A.;Elliott,T.S.;Townsley,F.M.;Pisa,R.;Davis,L.;S.J.;Ernst,R.J.;Lang,K.;Sachdeva,A.;Chin,J.W.Under Review.
(26)Yu,Z.;Pan,Y.;Wang,Z.;Wang,J.;Lin,Q.Angewandte Chemie International Edition2012,51,10600.
(27)Kamber,D.N.;Nazarova,L.A.;Liang,Y.;Lopez,S.A.;Patterson,D.M.;Shih,H.W.;Houk,K.N.;Prescher,J.A.J Am Chem Soc2013,135,13680.
(28)Wang,K.;Schmied,W.H.;Chin,J.W.Angew Chem Int Ed Engl2012,51,2288.
(29)Wang,K.;Neumann,H.;Peak-Chew,S.Y.;Chin,J.W.Nature biotechnology2007,25,770.
(30)Rackham,O.;Chin,J.W.Nature chemical biology2005,1,159.
(31)Deiters,A.;Schultz,P.G.Bioorganic&;Medicinal Chemistry Letters2005,15,1521.
- 还没有人留言评论。精彩留言会获得点赞!