卤素或含氮基团取代的联苄类似物及其制备方法和用途与流程
本发明属于药物合成领域,涉及一类联苄类似物及其制备方法和用途,具体涉及一类卤素或含氮基团取代的联苄类似物及其制备方法和用途。
背景技术:
恶性肿瘤常称癌症(cancer),是严重威胁人类健康的常见病、多发病。应用传统细胞毒类抗肿瘤药(antineoplastic drugs)或抗癌药(anticancer drugs)进行化疗在肿瘤的综合治疗(synthetic therapy)中占有重要地位。但化疗药物对占恶性肿瘤90%以上的实体瘤的治疗目前仍未能达到满意的疗效,肿瘤化疗存在两大主要障碍:包括药物的毒性反应和耐药性的产生。细胞毒类抗肿瘤药由于对肿瘤细胞缺乏足够的选择性,在杀伤肿瘤细胞的同时,对正常的组织细胞也产生不同程度的损伤作用,毒性反应成为肿瘤化疗时药物用量受限的关键因素。化疗过程中肿瘤细胞对药物产生不敏感现象即耐药性是肿瘤化疗失败的重要原因,亦是肿瘤化疗急需解决的难题。
近十几年来,随着肿瘤分子生物学和肿瘤药理学的理论和生物技术的不断发展,抗肿瘤药正从传统的细胞毒作用向针对机智的多环节作用的方向发展,其中新生血管生成抑制剂受到了越来越多的关注。
血管生成(angiogenesis)是指从已有的毛细血管或毛细血管后静脉发展而形成新的血管。肿瘤生长依赖于血管生成的概念始于20世纪70年代初,但其重要性并未受到关注。近二十年,由于发现了血管生成因子对血管生成的作用,以及血管生成对肿瘤生长和侵入转移、特别对肿瘤早期发生的重要影响,血管生成成为近年来肿瘤研究的热点之一,为肿瘤治疗开辟了一个新的思路。
联苄类化合物是指两个苯甲基单元通过甲基的碳碳单键相连而成的一类化合物。天然联苄具有抗肿瘤、植物生长调节、抑制血管新生等作用,但他们的结构相对单一、主要在于苯环上羟基与甲氧基取代位置和数量的变化。且这些天然联苄在植物中含量少,不容易大量获得。
技术实现要素:
本发明的目的在于提供一系列卤素或含氮基团取代的联苄类似物,并对这些化合物进行抗肿瘤活性测试和抑制血管新生测试,从而提供具有药用价值的联苄类似物新化学实体。
本发明的目的可通过以下措施来达到;
一种卤素或含氮基团取代的联苄类似物或其在药学上可接受的盐、水合物,所述的卤素或含氮基团取代的联苄类似物如式I所示:
其中,A为如式II所示的卤素或含氮基团取代的苯基或含1-2个N的五元或六元不饱和含氮杂环;
B、C、D可分别独立的选自氢、卤素、除甲氧基之外的烷氧基、硝基、胺基或取代胺基,但B、C、D不能同时为氢。
优选的,B、C、D可分别独立的选自氢、卤素或硝基,但B、C、D不能同时为氢。
进一步优选的,B、C分别独立的选自氢、卤素,D选自氢、卤素或硝基,但B、C、D不能同时为氢。
本发明所述的卤素为氟、氯、溴;所述的除甲氧基之外的烷氧基为C2-C5的烷氧基;所述的含1-2个N的五元或六元不饱和含氮杂环为2-吡啶基、3-吡啶基、4-吡啶基、嘧啶-5-基、嘧啶-2-基、吡咯-2-基、吡咯-3-基、吡唑-3-基、吡唑-4-基、咪唑-2-基、咪唑-4-基。
最优选的,本发明所述的卤素或含氮基团取代的联苄类似物自:4-羟基-3,5-二甲氧基-3’,4’-二氯联苄、4’-硝基-4-羟基-3,5-二甲氧基联苄、4-羟基-3,5-二甲氧基-4’-氯联苄、4-羟基-3,5-二甲氧基-2’-氯联苄、4-羟基-3,5-二甲氧基-4’-溴联苄、4-羟基-4’-氟-3,5-二甲氧基联苄、2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚、2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚、4-羟基-2’-氟-3,5-二甲氧基联苄、4-羟基-3,5-二甲氧基-3’-氟联苄、4-羟基-3,5-二甲氧基-2’,4’-二氯联苄、4-羟基-3,5-二甲氧基-2’,3’-二氯联苄。
本发明的另一目的在于提供一种卤素或含氮基团取代的联苄类似物的制备方法。
所述的卤素或含氮基团取代的联苄类似物的制备方法,包括:以4-羟基-3,5-二甲氧基苯甲醛为原料,与乙酸酐反应生成3,5-二甲氧基-4-乙酰氧基苯甲醛,通过硼氢化钠还原得到3,5-二甲氧基-4-乙酰氧基苯甲醇,再与二氯亚砜反应得到3,5-二甲氧基-4-乙酰氧基苯甲醇,进一步与三苯基膦反应生成(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷;(3,5-二甲氧基-4-乙酰 氧基苄基)-三苯基氯化磷与A-CHO反应生成相应的顺反混合的二苯乙烯再经甲醇钠酯交换得最后经氢气钯碳催化得目标化合物
其中,A为如式II所示的卤素或含氮基团取代的苯基或含1-2个N的五元或六元不饱和含氮杂环;
B、C、D可分别独立的选自氢、卤素、除甲氧基之外的烷氧基、硝基、胺基或取代胺基,但B、C、D不能同时为氢。
优选的,B、C、D可分别独立的选自氢、卤素或硝基,但B、C、D不能同时为氢。
进一步优选的,B、C分别独立的选自氢、卤素,D选自氢、卤素或硝基,但B、C、D不能同时为氢。
本发明中,所述的A-CHO具体为3,4-二氯苯甲醛、对硝基苯甲醛、对氯苯甲醛、邻氯苯甲醛、对溴苯甲醛、对氟苯甲醛、2-吡啶甲醛、4-吡啶甲醛、2-氟苯甲醛、3-氟苯甲醛、2,4- 二氯苯甲醛、2,3-二氯苯甲醛。
(1)、(2)、(3)三步反应产率高,达88.7%~99.8%,只需通过萃取将所得产物直接用于下一步反应,第(4)步反应产物会在体系中大量沉淀,通过抽滤得到白色纯品(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷。第(5)步反应为关键步骤,投料顺序和投料比例对反应成败至关重要,将(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷与A-CHO先溶于无水溶剂,充分搅拌,冰浴下缓慢加入缚酸剂氢化钠,其中(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷、A-CHO和氢化钠的摩尔比为1∶1∶1~1∶0.8∶1.2,(5)、(6)步反应只需通过萃取直接进入下一步反应,不需要柱色谱纯化;第(7)步反应结束通过硅胶柱色谱分离纯化得目标产物,洗脱剂为石油醚-乙酸乙酯(10∶1~5∶1V/V)混合溶剂或石油醚-丙酮(20∶1~3∶1)混合溶剂。
发明人对合成的卤素或含氮基团取代的联苄类似物进行体外五种癌细胞:结肠癌HCT116细胞、肺癌A549细胞、肝癌HepG2细胞、乳腺癌MDA-MB-231细胞和胃癌MKN-45细胞的细胞毒实验测试,表现出不同程度的抗癌作用,其中,对结肠癌HCT116具有最强的抗肿瘤活性,表明这类化合物具有显著的抑制肿瘤细胞增殖的作用。因此,本发明的另一个目的是提供所述的卤素或含氮基团取代的联苄类似物或其在药学上可接受的盐、水合物在制备治疗肿瘤药物中的应用。所述的肿瘤为结肠癌、肺癌、肝癌、乳腺癌、胃癌。
发明人对合成的卤素或含氮基团取代的联苄类似物以斑马鱼为模型,对它们进行抑制血管新生活性评价,体内斑马鱼血管新生抑制实验初筛结果表明这些化合物对斑马鱼血管新生表现出不同程度的抑制作用。进一步通过浓度梯度考察活性化合物对斑马鱼血管新生抑制作用,发现对斑马鱼呈现出浓度依赖性的血管新生抑制活性。这对于治疗与抗血管新生相关的肿瘤生长与转移、糖尿病视网膜疾病等具有重要意义。因此,本发明的另一个目的是提供所述的卤素或含氮基团取代的联苄类似物或其在药学上可接受的盐、水合物在制备抑制血管新生药物方面的应用。所述的抑制血管新生药物方面的应用包括在制备治疗抗血管新生相关的肿瘤生长与转移、糖尿病视网膜药物方面的应用。
本发明的有益效果:
本发明通过简单高效制备方法合成了一系列卤素或含氮基团取代的联苄类似物,以4-羟基-3,5-二甲氧基苯甲醛,先后经过乙酸酐乙酰基保护、硼氢化钠还原、二氯亚砜氯代和三苯基膦回流制得(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷关键中间体。关键中间体依次与不同取代的苯甲醛发生维悌希反应、甲醇钠酯交换反应、氢气钯碳还原;最终合成一系列目标产物。反应条件温和,容易重复,中间步骤不需要纯化处理,简化了实验过程,且总收率在32%~51%。这些合成的联苄类似物在抗肿瘤和抑制血管新生方面表现出不同程度的活性。 是潜在的抗肿瘤、治疗抗血管新生相关的肿瘤生长与转移、糖尿病视网膜等疾病的新化学实体,对于相关疾病新药的开发具有重要意义。
附图说明
图1为本发明血管转基因荧光斑马鱼受精后48小时体节间血管(ISVs)模型;其中,箭头指示体节间血管。图1A和图1A’为阴性对照试验;图1B-图1D和图1B’-图1D’是浓度分别为1.25μM,2.5μM和5μM的化合物4加药组斑马鱼试验;图1E和图1E’是阳性对照试验(2.5μM SU5416)。
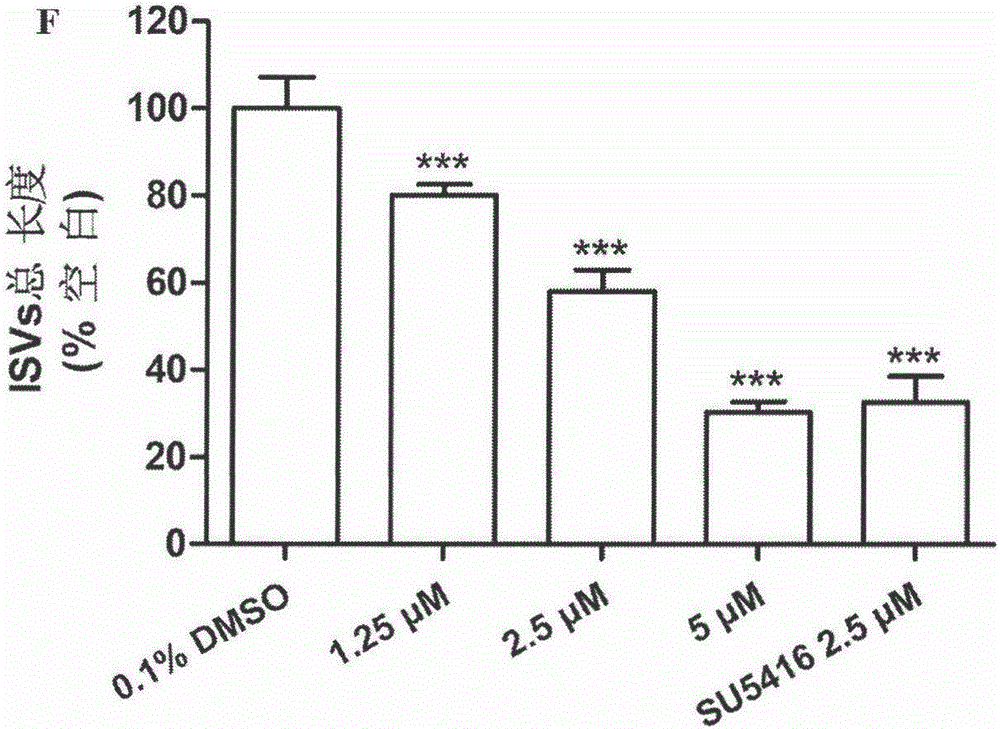
图2是斑马鱼受精后24小时加入化合物4作用24小时,即受精后48小时斑马鱼的体节间血管(ISVs)总长度统计图。
具体实施方式
结合具体实施例对本发明的技术方案做进一步详细的说明,但本发明并不限于以下实施例。
实施例1 4-羟基-3,5-二甲氧基-3’,4’-二氯联苄(化合物1)的制备
a、3,5-二甲氧基-4-乙酰氧基苯甲醛
在100mL单颈瓶中加入丁香醛(4-羟基-3,5-二甲氧基苯甲醛)粉末8.33g(0.046mol),二氯甲烷10mL,催化计量DMAP(4-二甲氨基吡啶),搅拌使充分溶解。缓慢滴加乙酸酐4.75mL(0.051mol),室温反应1h。加水洗去DMAP和乙酸酐,共洗3次,每次用水10mL。无水Na2SO4干燥有机层,浓缩回收二氯甲烷,得白色结晶10.19g,产率99.8%。经鉴定为3,5-二甲氧基-4-乙酰氧基苯甲醛。
ESI-MS m/z:225[M+H]+,相对分子质量224。
1H-NMR(300MHz,CDCl3)δ9.93(1H,s,-CHO),7.17(2H,s,H-2,H-6),3.92(6H,s,2×-OCH3),2.38(3H,s,-COCHx)。
b、3,5-二甲氧基-4-乙酰氧基苯甲醇
在100mL单颈瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲醛8.6g(0.038mol),乙醇10mL,充分搅拌。冰浴下缓慢加入NaBH4530mg(0.014mol),室温反应0.33h。滴加5%HCl调pH 至弱酸,减压回收溶剂,残渣加10mL水混悬,用二氯甲烷萃取3次,每次10mL。合并有机层,无水Na2SO4干燥,浓缩二氯甲烷,得白色固体8.34g,产率96.5%。经鉴定为3,5-二甲氧基-4-乙酰氧基苯甲醇。
ESI-MS m/z:227[M+H]+,相对分子质量226。
1H-NMR(300MHz,CDCl3)δ6.66(1H,s,H-2,H-6),4.69(2H,s,Ar-CH2-),3.84(6H,s,2×-OCH3),2.36(3H,s,-COCH3)。
c、3,5-二甲氧基-4-乙酰氧基苯甲基氯
在100mL单颈瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲醇8.3g(0.037mol),二氯甲烷10mL,搅拌溶解。冰盐浴下加入Et3N 6.2mL(0.043mol),SOCl23mL(0.043mol),室温反应3h。加入冰水停止反应,用二氯甲烷萃取3次,每次10mL。合并有机层,无水Na2SO4干燥,减压蒸干二氯甲烷,得淡黄色油状物7.96g,产率88.7%。经鉴定为3,5-二甲氧基-4-乙酰氧基苯甲基氯。
ESI-MS m/z:245[M-H]-,247[M+H]+,相对分子质量246。
1H-NMR(300MHz,CDCl3)δ6.85(1H,s,H-2,H-6),4.73(2H,s,Ar-CH2-),3.76(6H,s,2×-OCH3),2.24(3H,s,-COCH3)。
d、(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷
在100mL单颈瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲基氯7.8g(0.032mol),三苯基膦8.4g(0.032mol),甲苯5mL,搅拌溶解,加热回流10h。反应液降至室温,减压抽滤,用甲苯洗涤滤饼,干燥得白色粉末14.63g,产率90.2%。经鉴定为(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷。
ESI-MS m/z:541.2[M+Cl]-,相对分子质量506.5。
1H-NMR(300MHz,DMSO-d6)δ7.92(3H,m,ArH),7.76(6H,dt,J=3.6Hz,8.1Hz,ArH),7.69(6H,m,ArH),6.30(2H,d,J=2.4Hz,H-2,H-6),5.09(2H,J=15Hz,Ar-CH2-),3.37(6H,s,2×-OCH3),2.21(3H,s,×-OCH3)。
e、4-羟基-3,5-二甲氧基-3’,4’-二氯联苄(化合物1)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),3,4-二氯苯甲醛69mg(0.395mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点即顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-3’,4’-二氯二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-3’,4’-二氯二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=10∶1),得淡黄色结晶67mg,产率51%。经鉴定为4-羟基-3,5-二甲氧基-3’,4’-二氯联苄。
ESI-MS m/z:349.0[M+Na]+,325.1[M-H]-,相对分子质量为326。
1H-NMR(300MHz,CDCl3)δ7.34(1H,d,J=8.1Hz,H-5’),7.27(1H,brs,H-2’),6.98(1H,dd,J=1.8Hz,8.1Hz,H-6’),6.35(1H,s,H-2,H-6),5.41(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.84(4H,m,ArCH2CH2Ar)。
实施例2 4’-硝基-4-羟基-3,5-二甲氧基联苄(化合物2)的制备
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),对硝基苯甲醛60mg(0.397mmol)。氩气保护下加入无水THF 3mL,搅拌呈淡黄色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点即顺反异构二苯乙烯((Z/E)-4’-硝基-3,5-二甲氧基-4-乙酰氧基二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点 完全消失,证实脱乙酰基完全((Z/E)-4’-硝基-4-羟基-3,5-二甲氧基二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=5∶1),得亮黄色油状物46mg,产率39%。经鉴定为4’-硝基-4-羟基-3,5-二甲氧基联苄。
ESI-MS m/z:304.1[M+H]+,相对分子质量303。
1H-NMR(300MHz,CDCl3)δ8.15(2H,d,J=8.7Hz,H-3’,H-5’),7.31(2H,d,J=8.7Hz,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.0(4H,m,ArCH2CH2Ar)。
实施例3 4-羟基-3,5-二甲氧基-4’-氯联苄(化合物3)的制备
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),对氯苯甲醛56mg(0.398mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-氯二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-4’-氯二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=10∶1),得黄色固体41mg,产率36%。经鉴定为4-羟基-3,5-二甲氧基-4’-氯联苄。
ESI-MS m/z:315.1[M+Na]+,相对分子质量为292。
1H-NMR(300MHz,CDCl3)δ7.25(2H,d,J=8.4Hz,H-3’,H-5’),7.09(2H,d,J=8.1Hz,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.43(1H,s,ArOH),3.85(6H,s,2×-OCH3),2.86(4H,m,-CH2CH2-)。
实施例4 4-羟基-3,5-二甲氧基-2’-氯联苄(化合物4)的制备
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),邻氯苯甲醛50μL(0.427mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’-氯二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-2’-氯二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=10∶1),得白色固体54mg,产率47%。经鉴定为4-羟基-3,5-二甲氧基-2’-氯联苄。
ESI-MS m/z:315.1[M+Na]+,291.1[M-H]-,相对分子质量为292。
1H-NMR(300MHz,CDCl3)δ7.38(1H,m,H-3’),7.18-7.11(3H,m,H-4’,5’,6’),6.39(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.87(6H,s,2×-OCH3),3.05-2.84(4H,m,-CH2CH2-)。
实施例5 4-羟基-3,5-二甲氧基-4’-溴联苄(化合物5)的制备
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),对溴苯甲醛73mg(0.395mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-溴二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点 完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-4’-溴二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=10∶1),得黄色油状物42mg,产率32%。经鉴定为4-羟基-3,5-二甲氧基-4’-溴联苄。
ESI-MS m/z:359.0[M+Na]+,335.1[M-H]-,相对分子质量为336。
1H-NMR(300MHz,CDCl3)δ7.42-7.02(4H,m,ArH),6.37(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.86(6H,s,2×-OCH3),2.95-2.85(4H,m,-CH2CH2-)。
实施例6 4-羟基-4’-氟-3,5-二甲氧基联苄(化合物6)的制备
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),对氟苯甲醛43μL(0.402mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-氟二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-4’-氟二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶乙酸乙酯=10∶1),得淡黄色固体38mg,产率35%。经鉴定为4-羟基-4’-氟-3,5-二甲氧基联苄。
ESI-MS m/z:299.1[M+Na]+,275.1[M-H]-,相对分子质量为276。
1H-NMR(300MHz,CDCl3)δ7.10(2H,m,H-3’,H-5’),6.97(2H,m,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.85(6H,s,2×-OCH3),2.85(4H,m,-CH2CH2-)。
实施例7 4-羟基-2’-氟-3,5-二甲氧基联苄(化合物7)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),邻氟苯甲醛45μL(0.428mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’-氟二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-2’-氟二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=20∶1),得黄色油状物36mg,产率33%。经鉴定为4-羟基-2’-氟-3,5-二甲氧基联苄。
ESI-MS m/z:299.1[M+Na]+,相对分子质量为276。
1H-NMR(300MHz,CDCl3)δ7.21-7.01(4H,m,ArH),6.38(2H,s,H-2,H-6),5.40(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.86(4H,m,-CH2CH2-)。
实施例8 4-羟基-3,5-二甲氧基-3’-氟联苄(化合物8)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),间氟苯甲醛45μL(0.424mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-3’-氟二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实反应脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-3’-氟二苯乙烯)。滴加10%稀 盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=20∶1),得黄色油状物44mg,产率37%。经鉴定为4-羟基-3,5-二甲氧基-3’-氟联苄。
ESI-MS m/z:299.1[M+Na]+,相对分子质量为276。
1H-NMR(300MHz,CDCl3)δ7.23(1H,m,H-2’),6.95-6.88(3H,m,ArH),6.36(2H,s,H-2,H-6),5.43(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.87(4H,m,-CH2CH2-)。
实施例9 4-羟基-3,5-二甲氧基-2’,4’-二氯联苄(化合物9)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),2,4-二氯苯甲醛69mg(0.394mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’,4’-二氯二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实反应脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-2’,4’-二氯二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=10∶1),得黄色油状物52mg,产率40%。经鉴定为4-羟基-3,5-二甲氧基-2’,4’-二氯联苄。
ESI-MS m/z:349.0[M+Na]+,相对分子质量为326。
1H-NMR(300MHz,CDCl3)δ7.39(1H,d,J=2.1Hz,H-3’),7.14(1H,dd,J=1.8,8.1Hz,H-5’),7.03(1H,d,J=8.4Hz,H-6’),6.36(2H,s,H-2,H-6),5.43(1H,brs,ArOH),3.86(6H,s,2×-OCH3),3.01-2.80(4H,m,-CH2CH2-)。
实施例10 4-羟基-3,5-二甲氧基-2’,3’-二氯联苄(化合物10)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),2,3-二氯苯甲醛69mg(0.394mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’,3’-二氯二苯乙烯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实反应脱乙酰基完全((Z/E)-4-羟基-3,5-二甲氧基-2’,3’-二氯二苯乙烯)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=10∶1),得黄色油状物42mg,产率32%。经鉴定为4-羟基-3,5-二甲氧基-2’,3’-二氯联苄。
ESI-MS m/z:349.0[M+Na]+,325.2[M-H]-,相对分子质量为326。
1H-NMR(300MHz,CDCl3)δ7.34(1H,dd,J=1.5,7.8Hz,H-4’),7.10(1H,t,J=7.8Hz,H-5’),7.01(1H,dd,J=1.5,7.5Hz,H-6’),5.43(1H,brs,ArOH),3.87(6H,s,2×-OCH3),3.08-2.83(4H,m,-CH2CH2-)。
实施例11 2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚(化合物11)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),2-吡啶甲醛40μL(0.420mmol)。氩气保护下加入无水THF 3mL,搅拌呈亮黄色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成一个新的斑点推测为顺反异构((Z/E)-2,6-二甲氧基-4-(2-(吡啶-2-基)乙烯基)苯基乙酸酯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点 完全消失,证实反应脱乙酰基完全((Z/E)-2,6-二甲氧基-4-(2-(吡啶-2-基)乙烯基)苯酚)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=3∶1),得黄色结晶38mg,产率37%。经鉴定为2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚。
ESI-MS m/z:282.1[M+Na]+,258.1[M-H]-,相对分子质量为259。
1H-NMR(300MHz,CDCl3)δ8.57(1H,d,J=3.9Hz,H-3’),7.57(1H,dt,J=1.8,7.8Hz,H-4’),7.12(1H,m,H-5’),7.08(1H,d,J=7.8Hz,H-6’),6.39(2H,s,H-3,H-5),5.89(1H,brs,ArOH),3.81(6H,s,2×-OCH3),3.10-2.94(4H,m,-CH2CH2-)。
实施例12 2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚(化合物12)
在50mL单颈瓶中加入(3,5-二甲氧基-4-乙酰氧基苄基)-三苯基氯化磷200mg(0.395mmol),4-吡啶甲醛40μL(0.425mmol)。氩气保护下加入无水THF 3mL,搅拌呈白色混悬液。冰盐浴下缓慢加入NaH 11mg(0.458mmol),搅拌过夜。TLC监测生成两个新的斑点推测为顺反异构点((Z/E)-2,6-二甲氧基-4-(2-(吡啶-4-基)乙烯基)苯基乙酸酯),浓硫酸乙醇显色呈淡紫色,证明反应成功。加冰水终止反应,二氯甲烷萃取三次,有机层无水Na2SO4干燥,减压浓缩备用。
将浓缩物加5mL甲醇溶解,加入适量CH3ONa,反应0.5h。点板监测反应,顺反异构点完全消失,证实反应脱乙酰基完全((Z/E)-2,6-二甲氧基-4-(2-(吡啶-4-基)乙烯基)苯酚)。滴加10%稀盐酸调pH值至中性,减压浓缩溶剂。加入10mL水混悬,用二氯甲烷萃取三次,每次10mL,有机层无水Na2SO4干燥,减压浓缩。将浓缩物用5mL甲醇溶解,加入催化剂量10%Pd/C,通入氢气反应48h。抽滤得澄清甲醇溶液,即含目标化合物的粗品。用硅胶柱色谱纯化(石油醚∶丙酮=3∶1),得白色固体36mg,产率35%。经鉴定为2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚。
ESI-MS m/z:282.1[M+Na]+,相对分子质量为259。
1H-NMR(300MHz,CDCl3)δ8.50(2H,d,J=5.7Hz,H-3’,5’),7.09(2H,d,J=4.0Hz,H-2’,6’),6.33(2H,s,H-3,H-5),5.31(1H,brs,ArOH),3.84(6H,s,2×-OCH3),2.89(4H,m,-CH2CH2-)。
表1 本发明制得的卤素或含氮基团取代的联苄类似物
实施例14 本发明化合物体外抑制肿瘤细胞增殖实验(MTT法)
1、材料
(1)细胞株及试剂
人肺癌细胞株A549、人肝癌细胞株HepG2、人胃癌细胞株MKN-45、乳腺癌细胞株MDA-MB-231、结肠癌细胞株HCT116均购自中国科学院上海生命科学研究院细胞库;
高糖DMEM培养液购自美国Gibco公司;
胎牛血清购自杭州四季青公司;
胰酶细胞消化液购自上海碧云天生物技术有限公司;
噻唑蓝(MTT)购自sigma公司。
PBS缓冲盐溶液:磷酸二氢钾(KH2PO4)0.27g,磷酸氢二钠(Na2HPO4)1.42g,氯化钠(NaCl)8g,氯化钾(KCl)0.2g,加去离子水约800mL充分搅拌溶解,然后加入浓盐酸调pH至7.4, 最后定容到1L,高温高压灭菌后置于4摄氏度冰箱保存待用。
(2)受试药品溶液
化合物1-12用DMSO溶解配制成25mM的溶液,用高糖DMEM培养液稀释为250μM的母液,在此基础上依次用高糖DMEM培养液稀释得25,5,1,0.2,0.04μM的受试药品溶液,4℃保存备用。
(3)细胞培养
人肺癌细胞A549、人肝癌细胞HepG2、人胃癌细胞MKN-45、乳腺癌细胞MDA-MB-231、结肠癌细胞HCT116在含10%胎牛血清的DMEM培养液中,37℃、5%CO2保持饱和湿度培养,2-3天传代一次。
分别取对数生长期癌细胞一瓶,加入胰酶细胞消化液消化,使贴壁细胞脱落,配制成浓度为5×104个/mL的细胞悬液,96孔细胞培养板中每孔加入100μL细胞悬液(每孔5×103个细胞),板置于37℃,5%CO2培养箱中培养24小时。弃去培养液,PBS缓冲盐溶液清洗2次,每孔加入100μL相应的含不同浓度受试药品溶液的高糖DMEM培养液;同时设立阴性对照组(只加高糖DMEM培养液)。
96孔板置于37℃,5%CO2培养箱中培养72小时,每孔加入10μL MTT溶液(5mg/mL),在培养箱继续培养4小时。弃去培养基,每孔加入150μL DMSO溶解,摇床10分钟轻轻混匀;每孔的吸光度(OD值)用1500酶标仪在490nm波长下进行检测,计算抑制率和IC50值。
应用GraphPad Prism 5软件计算IC50。
表2 化合物1-12对癌细胞的细胞毒活力测试结果
由表2可知,化合物1-12对结肠癌HCT116细胞、肺癌A549细胞、肝癌HepG2细胞、乳腺癌MDA-MB-231细胞和胃癌MKN-45细胞表现出不同程度的抗癌作用,其中,对结肠癌HCT116具有最强的抗肿瘤活性。结果表明本发明联苄类似物具有显著的抑制肿瘤细胞增殖的作用。
实施例15 以转基因斑马鱼为模型评价本发明化合物抗血管新生抑制活性
1、材料
(1)实验动物
转基因斑马鱼Tg(flil:EGFP)购自南京大学模式生物,成鱼鱼龄:6个月-9个月;饲养光照条件:14h/10h明暗交替,温度28.5℃,pH 7.2-7.6。实验全程均使用转基因斑马鱼幼体胚胎(受精后24小时)作为实验对象。
(2)药物与试剂
本发明合成的卤素或含氮基团取代的联苄类似物化合物1-12;
红十字小丑盐(Instant Ocean salt):Instant Ocean公司;
二甲基亚砜(DMSO):美国Sigma-Aldrich公司;
链霉蛋白酶(Pronase):美国Sigma公司;
三卡因(Tricaine):美国Sigma公司;
VEGFR(血管内皮细胞生长因子受体)特异性血管新生抑制剂SU5416:美国Sigma公司。
(3)主要实验仪器
倒置荧光显微镜(Olympus IX71);
超纯水机(Milli-Q PLUS 185);
恒温培养箱:上海知楚仪器有限公司;
体视显微镜(Nikon SMZ745)。
2、主要实验溶液和药物的配制
卵水(Egg water)配制方法:1L去离子水加入0.2g红十字小丑盐(Instant Ocean salt);
0.1%DMSO溶液(阴性对照)配制:使用时用卵水配制成浓度为0.1%的工作液,现配 现用;
VEGFR(血管内皮细胞生长因子受体)特异性血管新生抑制剂SU5416作为本组实验阳性对照药,使用时用0.1%DMSO溶液配制成实验所需的浓度,阳性对照药浓度为2.5μM;12个卤素或含氮基团取代的联苄类似物的血管新生抑制初筛浓度均为50μM,有抑制活性的化合物再用0.1%DMSO溶液稀释寻找化合物抑制血管新生的最低有效剂量范围。
3、方法
本实验材料为发育正常的受精后24小时龄转基因斑马鱼(可在血管内皮细胞特异性表达绿色荧光蛋白)。使用链霉蛋白酶(0.5mg/mL)将其卵膜消化除去得到脱去卵膜的斑马鱼胚胎。以20个斑马鱼胚胎/孔将斑马鱼随机分配至含1mL培养液的24孔板内,并立即向孔内培养液中加入对应浓度的化合物溶液,阴性对照组加入等量的0.1%DMSO溶液,加药完毕后将24孔板转移至28.5℃恒温生物培养箱中。继续培养24小时(即胚胎发育至受精后48小时),取出孔板,使用倒置荧光显微镜观察,拍摄血管荧光照片并对斑马鱼血管新生的情况进行统计。
体内斑马鱼血管新生抑制实验结果见表3,大部分化合物表现出不同程度的血管新生抑制活性,其中化合物4的抑制斑马鱼血管新生活性强于阳性药。
以化合物4为代表,图1表示斑马鱼体节间血管生成的抑制率随着化合物4浓度的上升而呈现梯度增加,即化合物对斑马鱼血管新生呈现出浓度依赖性的抑制活性。图2为化合物4对斑马鱼体节间血管生长抑制作用的统计图。
表3 化合物1-12对斑马鱼血管新生抑制活性
- 还没有人留言评论。精彩留言会获得点赞!