一类新颖结构倍半萜化合物及其在制备抗炎药物中的应用的制作方法
本发明属于属于天然产物领域,具体涉及两个结构新颖的倍半萜类化合物及其在制备抗炎药物中的应用。
背景技术:
:
新颖结构天然产物的持续发现是药物研发的重要源泉之一。目前,从极端环境微生物,特别是从南极来源真菌中,获得骨架新颖的活性次生代谢产物已逐渐成为天然药物研究的重要方向之一。在对南极来源真菌的次生代谢产物的研究过程中,我们分离获得两个新颖骨架的倍半萜类化合物,它们具有很强的抗炎作用,且在有效剂量下,未发现有细胞毒性。因此是开发成为结构新颖、活性高效、低毒副作用的抗炎药物的理想侯选化合物。
技术实现要素:
:
本发明的第一个目的的是提供两个具有抗炎活性的新骨架倍半萜类化合物及其药用盐。
本发明的两个倍半萜类化合物或其药用盐,其结构如式(Ⅰ)和(Ⅱ)所示:
化合物1的结构如(Ⅰ)所示,化合物2的结构如式(Ⅱ)所示。
本发明的第二个目的是上述倍半萜类化合物的制备方法,所述的倍半萜类化合物是从曲霉(Aspergillus sp.)SCSIO 05702的发酵培养物中制备分离得到的。
优选,具体步骤如下:
a、制备曲霉(Aspergillus sp.)SCSIO 05702的发酵培养物;
b、将发酵培养物的发酵液和菌丝体分离,发酵液用乙酸乙酯萃取,合并乙酸乙酯萃取液,浓缩后得到浸膏A;菌丝体先用丙酮浸提,合并浸提液,浸提液回收丙酮后剩余水混合液用乙酸乙酯萃取,乙酸乙酯萃取液浓缩后得到浸膏B,将浸膏A和浸膏B合并,得到粗提物;粗提物经中压正相硅胶柱层析,二氯甲烷/甲醇从100:0梯度洗脱至0:100,收集二氯甲烷/甲醇体积比为95:5洗脱的馏分;将该馏分进行葡聚糖凝胶Sephadex LH-20层析纯化处理,以二氯甲烷/甲醇体积比1:1作为流动相洗脱,收集馏分再经纯化后得到如式(Ⅰ)和式(Ⅱ)所示的化合物。
所述的a步骤的制备曲霉(Aspergillus sp.)SCSIO 05702的发酵培养物优选通过以下方法制备:将活化的曲霉(Aspergillus sp.)SCSIO 05702接入种子培养基中,25℃,180rpm,培养72h制得种子液,将种子液以体积百分比5%的接种量接入到发酵培养基中,25℃,摇床培养35d,而制得发酵培养物,所述的种子培养基和发酵培养基的配方为每升培养基中含有:甘露醇20g、麦芽糖20g、葡萄糖10g、谷氨酸钠10g、KH2PO4 0.5g、MgSO4·7H2O 0.3g、酵母浸膏3g、玉米干粉0.3g、余量为水,pH 7.5。按上述组份和含量混合均匀,然后121℃,灭菌30min。
本发明的第三个目的是提供曲霉(Aspergillus sp.)SCSIO 05702在制备上述如式(Ⅰ)或式(Ⅱ)所示的倍半萜类化合物中的应用。
本发明通过实验发现,化合物1和2对脂多糖(LPS)诱导小鼠巨噬细胞株RAW264.7产生的一氧化氮具有较强的抑制作用,在浓度为25μM时,其抑制率分别为70.1%和67.1%,在相同浓度下,对RAW264.7细胞无细胞毒活性,因此可望开发成为无毒的抗炎新药。
因此,本发明的第四个目的是提供如式(Ⅰ)或式(Ⅱ)所示的倍半萜类化合物在制备抗炎药物中的应用。
综上可知,本发明的曲霉(Aspergillus sp.)SCSIO 05702能够产生具有抗炎活性的新颖倍半萜类化合物,该类化合物对LPS诱导小鼠巨噬细胞株RAW264.7释放NO具有较强的抑制作用,因此能够用于制备抗炎药物。
本发明为制备具有抗炎活性的一类结构新颖的倍半萜类化合物的生产制备提供了一种新的方法,为开发高效、低毒的抗炎药物提供了理想的侯选化合物。
本发明的曲霉(Aspergillus sp.)SCSIO 05702于2015年01月06日保藏于中国微生物菌种保藏管理委员会普通微生物中心(CGMCC),地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,保藏编号为:CGMCC No.10279。
附图说明:
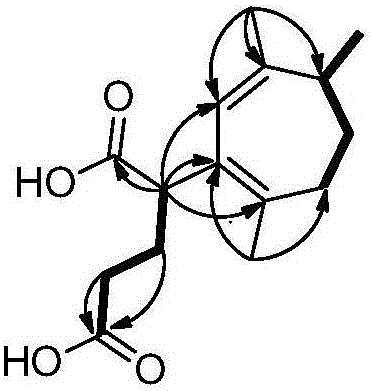
图1是化合物1关键的1H–1H COSY(加粗黑线)和HMBC(单箭头)相关;
图2是化合物2关键的1H–1H COSY(加粗黑线)和HMBC(单箭头)相关;
图3是化合物1和2的实测CD及计算ECD图谱。
具体实施方式:
以下实施例是对本发明的进一步说明,而不是对本发明的限制。
实施例1:如式(Ⅰ)所示的化合物1和如式(Ⅱ)所示的化合物2的制备和结构鉴定
一、如式(Ⅰ)所示的化合物1和如式(Ⅱ)所示的化合物2的制备
1、种子培养基(发酵培养基):按每升培养基中重量计算,含有甘露醇20g,麦芽糖20g,葡萄糖10g,谷氨酸钠10g,KH2PO4 0.5g,MgSO4·7H2O 0.3g,酵母浸膏3g,玉米干粉0.3g,余量为水,pH 7.5。每升是这样配制的:将甘露醇20g,麦芽糖20g,葡萄糖10g,谷氨酸钠10g,KH2PO4 0.5g,MgSO4·7H2O 0.3g,酵母浸膏3g,玉米干粉0.3g加入到水中,然后用水定容至1L,调pH值至7.5,然后121℃,灭菌30min。
2、发酵
2.1、种子培养:将活化的曲霉(Aspergillus sp.)SCSIO 05702接入每瓶含有50mL种子培养基的250mL的锥形培养瓶中,25℃,180rpm,培养72h制得种子液。
2.2、发酵培养:将种子液以5%的接种量(体积百分比)接入到30L发酵培养基中,25℃,摇床培养35d,而制得发酵培养物。
3、萃取:将发酵液和菌丝体分离,发酵液用乙酸乙酯萃取4次,合并乙酸乙酯萃取液,减压蒸馏浓缩后得到浸膏A;菌丝体先用丙酮超声提取3次,每次15min,合并浸提液,浸提液减压回收丙酮后剩余水混合液用乙酸乙酯萃取,乙酸乙酯萃取液减压蒸馏浓缩后得到浸膏B,将浸膏A和浸膏B合并,得到粗提物(28.6g)。
4、化合物1和化合物2的分离
将浸膏A和浸膏B合并的粗提物(28.6g)正相硅胶柱层析,二氯甲烷/甲醇从100:0梯度洗脱至0:100,收集二氯甲烷/甲醇体积比为95:5洗脱的馏分,进一步经葡聚糖凝胶Sephadex LH-20柱子,采用二氯氯仿-甲醇(体积比1:1)洗脱,洗脱得到的馏分命名为洗脱物;
上述洗脱物采用高效液相HPLC进行精细分离,洗脱体系为:甲醇/水(体积比60:40),柱子:YMC-pack ODS-A,10×250mm,5μm,流速为4mL/min,得到化合物1(20.5mg,保留时间是26.1min)。
洗脱物采用高效液相HPLC进行精细分离,洗脱体系为:乙腈/水(体积比38:62),柱子:YMC-pack ODS-A,10×250mm,5μm,流速4mL/min,得到化合物2(9.5mg,保留时间是22.2min)。
二、化合物1和化合物2的结构鉴定
对化合物1和化合物2进行结构分析测试,得到以下理化性质数据:
化合物1:淡黄色油状;[α]25D-87.8(c 1.0,MeOH);UV(MeOH)λmax(logε)202(4.13)nm;CD(0.2μg/mL,MeOH),λmax(Δε)207(-11.22),225(-5.48),and 245(-14.49)nm;1H和13C NMR数据见表1;(–)-HRESIMS m/z 251.1645[M–H]–(calcd for C15H23O3,251.1653).
化合物2:淡黄色油状;[α]25D-91.9(c 0.95,MeOH);UV(MeOH)λmax(logε)203(4.26),376(3.40)nm;CD(0.2μg/mL,MeOH),λmax(Δε)207(-7.28),228(-1.21),and 247(-9.68)nm;1H和13C NMR数据见表1;(–)-HRESIMS m/z 265.1450[M–H]–(calcd for C15H21O4,265.1445).
表1.化合物1和化合物2的1H-和13C-NMR数据归属。
在Bruker Avance 500NMR色谱仪上测量所得,溶剂为CD3OD。
化合物1关键的1H–1H COSY(加粗黑线)和HMBC(单箭头)相关如图1所示,化合物2关键的1H–1H COSY(加粗黑线)和HMBC(单箭头)相关如图2所示。化合物1和2的实测CD及计算ECD图谱如图3所示。
由此鉴定化合物1的结构如式(Ⅰ)所示,化合物2的结构如式(Ⅱ)所示。
实施例3化合物1和化合物2的抗炎活性测定
化合物1和化合物2对RAW264.7巨噬细胞中脂质过氧化酶(LPS)诱导NO生成具有抑制活性,实验方法参考文献(Wang,J.F.;Dong,S.S.;Wang,Y.H.;Lu,Q.;Zhong,H.M.;Du,G.H.;Zhang,L.;Cheng,Y.X.Cyclic diarylheptanoids from Myrica nana inhibiting nitric oxide release.Bioorg.Med.Chem.2008,16,8510–8515),活性结果所示:在25μM的浓度下,化合物1和化合物2表现出很强的抑制活性,其抑制率分别为70.1%和67.1%。相同剂量下,阳性对照药地塞米松的抑制率为92.6%。此外,在25μM的浓度下,化合物1和化合物2没有表现出细胞毒活性,因此提示能够被用于制备低毒高效的抗炎药物。
表2.化合物1和化合物2对LPS-诱导的NO生成的抑制活性结果
- 还没有人留言评论。精彩留言会获得点赞!