大丽轮枝菌VdKu80基因敲除突变体菌株,其构建方法及其应用与流程
本发明涉及大丽轮枝菌突变体菌株及其应用,特别涉及敲除VdKu80基因的大丽轮枝菌突变体菌株,还涉及以所述大丽轮枝菌突变体菌株为受体菌株进行基因定点敲除的应用,属于真菌分子生物学领域。
背景技术:
大丽轮枝菌(Verticillium dahliae Kleb.)是一种重要的植物病原真菌,属于子囊菌门、粪壳菌科、轮枝菌属。它在全世界范围内有200种以上的寄主,包括乔木、灌木、经济作物和观赏性花卉等。由其引起的病害会导致植物枯萎死亡,因此造成了巨大的经济损失。另外,大丽轮枝菌也是导致北京香山黄栌枯萎病的病原真菌,每年造成大量的黄栌枯萎死亡,严重的影响了香山红叶的生态价值和景观价值。对于由大丽轮枝菌引起的枯萎病,目前主要防治措施包括a.物理方法:加强营林措施、施肥灌溉增强树势、及时清理病害木残体防止发生再次侵染;b.化学方法:使用各种农药灌根、直接注射至受害植物以及使用化学农药熏蒸处理土壤;c.生物方法:筛选对大丽轮枝菌具有拮抗作用的真菌(木霉)和细菌(枯草芽孢杆菌)、选育抗病植物。但是目前使用的这些方法防治效果并不理想。导致其难以防治主要有两个原因:1、大丽轮枝菌主要危害寄主的维管束,一般的化学农药以及拮抗菌无法发挥功能,因此效果大打折扣;2、大丽轮枝菌在其病害循环的后期会形成黑色素化的休眠结构“微菌核”,该结构由膨胀分隔的菌丝聚集而成,对于不良的外界环境具有极强的抗逆性,并且能够在土壤内存活数年以上,一旦遇到合适的寄主,微菌核就会萌发形成菌丝,作为初侵染源,继续对植物进行危害。
随着科学技术的发展,人们开始大量研究大丽轮枝菌致病的分子机制以及大丽轮枝菌微菌核形成的分子机制,从而为大丽轮枝菌的防治提供理论依据。这方面的工作在农业有害病原真菌稻瘟菌(Magnaporthe oryzae)中取到了较大进展,大量参与附着胞形成及侵染过程的相关基因功能被研究,明确了它们在稻瘟菌侵染及致病的过程中发挥的功能并解析了它们之间的调控网络,为稻瘟菌的防治提供了巨大的价值。因此,通过基因功能研究揭示大丽轮枝菌致病分子机制和微菌核形成分子机制,对于病害的防治意义重大。
对于大丽轮枝菌的基因功能研究,近十年来取得到了巨大的进展,尤其是在大丽轮枝菌全基因序列公布之后。大丽轮枝菌共有10535个编码基因,而且随着研究的进一步深入,为了更好的解析大丽轮枝菌的致病及微菌核形成的分子机制,开展高通量的基因功能研究是十分必要的。但是目前国内外已经研究功能的基因数量还非常有限,造成这一现象的一个重要原因是其同源重组率低,导致基因敲除效率不高。虽然近年来,研究者通过改变载体构建方法从double joint PCR到Split-marker、改变转化方法从PEG介导的原生质体转化到农杆菌介导的转化等都有效的提高了基因敲除效率,但是目前基因敲除效率依然是限制大丽轮枝菌基因功能研究的主要因素之一。因此,提高大丽轮枝菌基因敲除效率对加快大丽轮枝菌基因功能研究的进程具有重要的意义。
在真核生物中,细胞会通过同源重组(homologous recombination)和非同源末端连接(non-homologous end joining)等方式修复受到破坏的双链DNA,通常非同源末端连接方式是真核生物中主要的修复方式,它能抑制外源DNA在细胞内发生同源重组事件,因此该方式能有效的降低同源重组率,从而导致基因敲除效率降低。参与非同源末端连接修复方式的基因主要包括Ku70、Ku80、XRCC4、DNAPKcs和LIG4等,其中Ku70和Ku80组成的Ku异质二聚体负责识别受损的双链DNA,因此它们在非同源末端连接修复途径中发挥着重要的作用。大量的研究表明敲除Ku70或Ku80基因能够大大的提高真菌的基因敲除率,这一现象在米曲霉(Aspergillus oryzae)、稻瘟菌(M.oryzae)、灰葡萄孢(Botrytis cinerea)、粗糙脉胞菌(Neurospora crassa)、黄青霉(Penicillium chrysogenum)等很多真菌中得到了验证。已有研究显示,敲除大丽轮枝菌Ku70基因也能够显著提高基因敲除率,从原有的0.5%提高到了20%以上,因此这一机制在大丽轮枝菌中也同样适用。但是对于敲除Ku80基因能否显著提高其基因敲除率并没有研究。
故本发明旨在获得具有高效基因敲除效率的大丽轮枝菌菌株。本发明通过PEG介导的原生质体转化方法,成功敲除大丽轮枝菌VdKu80基因。结果表明△VdKu80突变体菌株其基因敲除率较野生型菌株得到了显著的提高,并且大丽轮枝菌△VdKu80突变体菌株在菌丝生长、产孢、胁迫响应、微菌核形成以及致病性方面与野生型菌株无显著差异。因此本发明获得的大丽轮枝菌△VdKu80突变体菌株可以用于后续的基因功能研究。该突变体菌株的使用将大大提高大丽轮枝菌基因功能研究效率。
技术实现要素:
本发明的目的是针对现有大丽轮枝菌存在基因同源重组率低,基因敲除率低的技术问题,提供一株大丽轮枝菌(Verticillium dahliae)XS11的VdKu80基因敲除突变体菌株、其构建方法以及应用。本发明方法构建的VdKu80基因敲除突变体菌株在敲除VdKu80基因后,其基因敲除效率显著提高,而且敲除VdKu80基因并不影响突变体菌株的菌丝生长、产孢、胁迫响应、微菌核形成和致病性。因此,敲除VdKu80基因的大丽轮枝菌突变体菌株能够作为受体菌株对大丽轮枝菌其他基因进行敲除实验。
为实现本发明的目的,本发明一方面提供一株大丽轮枝菌(Verticillium dahliae)XS11的VdKu80基因敲除突变体菌株VdXS11-△VdKu80,其微生物保藏号为CGMCC No.12873。
其中,所述VdKu80基因被潮霉素抗性基因所替代。
本发明另一方面提供一种大丽轮枝菌(Verticillium dahliae)的VdKu80基因敲除突变体菌株VdXS11-△VdKu80的构建方法,包括如下步骤:1)利用Split-marker方法构建大丽轮枝菌VdKu80基因敲除载体;2)利用PEG介导的方法,将VdKu80基因敲除载体导入野生型大丽轮枝菌的原生质体内,获得大丽轮枝菌VdKu80基因敲除的转化子;3)利用PCR方法对大丽轮枝菌VdKu80基因敲除转化子进行筛选。
其中,步骤1)中所述大丽轮枝菌VdKu80基因敲除载体的构建方法,包括如下步骤:
1A)以大丽轮枝菌基因组DNA为模板,分别以Ku80-5F/Ku80-5R、Ku80-3F/Ku80-3R为特异性引物进行PCR扩增,获得大丽轮枝菌VdKu80基因上游片段VdKu80 5F、下游片段VdKu80 3F;
1B)以质粒gGFP为模板,以Hyg-For/Hyg-Rev为特异性引物进行PCR扩增,获得潮霉素抗性基因片段Hygromycin片段;
1C)将VdKu80 5F、VdKu80 3F片段、Hygromycin片段分别与pMDTM19-T Vector质粒进行连接反应,获得VdKu80 5F重组质粒、VdKu80 3F重组质粒和Hygromycin重组质粒;
1D)以VdKu80 5F重组质粒为模板,以Ku80-5F/Ku80-5RO为引物,进行PCR扩增得到VdKu80 5FO片段;以VdKu80 3F重组质粒为模版,以Ku80-3FO/Ku80-3R为引物,进行PCR扩增得到VdKu80 3FO片段;以Hygromycin重组质粒为模版,以M13F/M13R为引物,进行PCR扩增得到HygromycinO片段;
1E)以VdKu80 5FO片段和HygromycinO片段为模板,以Ku80-5F/HY-R为引物,进行融合PCR扩增,获得大丽轮枝菌VdKu80基因敲除载体1:VdKu80 5F+2/3Hyg片段;以VdKu80 3FO片段和HygromycinO片段为模板,以YG-F/Ku80-3R为引物,进行融合PCR扩增,获得大丽轮枝菌Ku80基因敲除载体2:2/3Hyg+VdKu80 3F片段。
特别是,步骤1D)中VdKu80 5FO片段在3’端含有一段与M13F相同的DNA序列;VdKu80 3FO片段在5’端含有一段与M13R相同的DNA序列;HygromycinO片段的5’和3’分别含有M13F和M13R的DNA序列。
其中,步骤2)中所述野生型大丽轮枝菌菌株为大丽轮枝菌XS11菌株。
特别是,步骤2)中所述原生质体转化包括如下步骤:
2A)将野生型大丽轮枝菌分生孢子置于液体YEPD培养基中培养,得到大丽轮枝菌新鲜菌丝。
2B)利用Lyzing和Driselase酶,对大丽轮枝菌新鲜菌丝进行酶解,获得原生质体;
2C)将大丽轮枝菌VdKu80基因敲除载体VdKu80 5F+2/3Hyg片段和2/3Hyg+VdKu80 3F片段与大丽轮枝菌原生质体混合后,在一定浓度的PEG条件下导入大丽轮枝菌原生质体内,获得大丽轮枝菌VdKu80基因敲除的转化子。
其中,步骤3)中所述PCR扩增方法筛选大丽轮枝菌VdKu80基因敲除转化子包括如下步骤:
3A)使用CTAB法提取大丽轮枝菌VdKu80基因敲除转化子的DNA;
3B)以步骤3A)提取的DNA为模板,以Ku80-IF/Ku80-IR为引物,对所有转化子进行PCR扩增;
3C)对PCR扩增产物进行凝胶电泳检测,PCR扩增产物中未能扩增出Ku80基因片段(102bp大小)条带的转化子筛选为大丽轮枝菌VdKu80基因敲除突变体。
特别是,还包括步骤4),采用Southern blot方法对大丽轮枝菌VdKu80基因敲除转化子进行分析、鉴定。
其中,所述大丽轮枝菌VdKu80基因敲除转化子的分析、鉴定包括如下步骤:
4A)制备探针
以VdKu80 3F重组质粒为模板,以Ku80-3F/Ku80-P1为引物,进行PCR扩增获得探针片段;接着使用DIG High Prime DNA Labling and Detection Starter Kit I(Roche)试剂盒中的DIG-High Prime并按照试剂盒说明书对探针片段进行标记,制得地高辛标记探针;
4B)杂交处理
使用CTAB法提取大丽轮枝菌VdKu80基因敲除转化子的DNA;接着用KpnI限制性内切酶对DNA进行酶切处理;然后将酶切后的DNA片段经琼脂糖凝胶电泳分离后转移至N+的硝酸纤维素膜上;再利用地高辛(DIG)标记探针进行杂交。
特别是,还包括对杂交后的硝酸纤维素膜按照DIG High Prime DNA Labling and Detection Starter Kit I(Roche)试剂盒的要求进行洗膜,显色。
本发明又一方面提供一种大丽轮枝菌VdKu80基因敲除突变体菌株VdXS11-△VdKu80-9作为受体菌株在提高基因敲除效率中的用途。
本发明与现有技术相比,具有以下优点:
本发明通过PEG介导的原生质体转化方法,成功敲除大丽轮枝菌VdKu80基因,获得敲除Ku80基因的突变体菌株VdXS11-△VdKu80-9,该突变体菌株的基因敲除率较野生型菌株得到了显著的提高,基因敲除率提高20%以上;而且本发明的大丽轮枝菌△VdKu80突变体菌株在菌丝生长、产孢、胁迫响应、微菌核形成以及致病性方面与野生型菌株无显著差异。因此本发明获得的大丽轮枝菌△VdKu80突变体菌株可以用于后续的基因功能研究,并且能大大提高大丽轮枝菌基因功能研究效率。
附图说明
图1A是VdKu80基因上游、下游片段的电泳检测图;
图1B是VdKu80基因上游、下游片段分别融合2/3Hygromycin片段的电泳检测图;
图2A是利用Ku80-IF/Ku80-IR引物进行PCR验证VdKu80基因敲除突变体的电泳检测图;
图2B是利用Southern blot鉴定VdKu80基因敲除突变体的检测图;其中:
WT表示大丽轮枝菌野生型;△VdKu80_1、△VdKu80_4、△VdKu80_6、△VdKu80_9表示成功敲除了VdKu80基因的敲除突变体;VdKu80_5表示构建的VdKu80敲除片段异位整合到基因组上其他位置上了。用于Southern blot分析的野生型和突变体基因组均使用KpnI限制性内切酶进行酶切。
图3A是大丽轮枝菌野生型菌株和△VdKu80突变体菌株在培养基PDA、CM、CM+100μg/ml Cong Red、CM+1M NaCl上生长的菌落直径统计结果图;
图3B是大丽轮枝菌野生型菌株和△VdKu80突变体菌株在培养基PDA、CM、CM+100μg/ml Cong Red、CM+1M NaCl上生长菌落形态图;
图3C是大丽轮枝菌野生型菌株和△VdKu80突变体菌株在培养基PDA上生长10天后的产孢量统计结果图。
图4是大丽轮枝菌野生型菌株和△VdKu80突变体菌株在BM培养基上培养5天后,微菌核形成的显微镜观察;
图5是大丽轮枝菌野生型菌株和△VdKu80突变体菌株对烟草幼苗致病性测定,其中:CK蒸馏水对照组,将烟草幼苗的根部浸泡在含106孢子/ml的野生型菌株和△VdKu80突变体菌株的孢子悬浮液中10分钟,对照使用蒸馏水。图片拍摄于接种后25天。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
下述实施例中的方法,无特别说明,均为常规方法。PCR反应中所用的DNA聚合酶、dNTP,提取基因组的试剂盒和PCR产物纯化的试剂盒等分子生物学常规试剂均购自宝生物工程(大连)有限公司和天根生化科技(北京)有限公司,相应的分子生物学操作按照商品试剂说明书和《分子克隆》(第三版)进行。
本发明所涉及的术语除另外定义之外,所使用的技术及术语均与本发明所属领域普通技术人员通常所了解的具有相同含义。
实施例1材料、试剂、培养基配制
1、野生型菌株和质粒
大丽轮枝菌XS11作为本发明使用的野生型菌株,采集自香山地区枯萎的黄栌病部组织。
质粒gGFP,用于扩增潮霉素基因片段,购自于美国堪萨斯州的真菌遗传储存中心(Fungal Genetics Stock Center)。
质粒pSilent-Dual1,用于扩增遗传霉素基因片段,购自于美国堪萨斯州的真菌遗传储存中心(Fungal Genetics Stock Center)。
质粒pMDTM19-T Vector,用于基因克隆,购自于宝生物工程(大连)有限公司。
2、培养基
2-1)PDA固体培养基
200g土豆,20g D-glucose(D-葡萄糖),15g琼脂粉,无菌水定容至1L,置于121℃,灭菌30min。
2-2)CM固体培养基
50ml 20×nitrate salts,1ml 1000x Trace,10g D-glucose,2g peptone(细菌蛋白胨),1g yeast extract(酵母浸膏),1g casamino acids(酸水解酪素),1ml vitamin solution,用NaOH调pH至6.5,无菌水定容至1L,琼脂粉15g/L,置于121℃,灭菌30min。
2-3)胁迫培养基1(含有100μg/ml Cong Red(刚果红)的CM固体培养基):
使用前每1L CM固体培养基中加入Cong Red 10mg(即Cong Red的浓度为100μg/ml)。
2-4)胁迫培养基2(含1M的NaCl的CM固体培养基)
使用前每1L CM固体培养基中加入58.5g的NaCl(即NaCl的浓度为1M)。
2-5)微菌核生长培养基(BM固体培养基)
10g D-glucose,0.2g sodium nitrate,0.52g KCl,0.52g MgSO4·7H2O,1.52g KH2PO4,3μM thiamine HCl,0.1μM biotin,15g琼脂粉,无菌水定容至1L,置于121℃,灭菌30min。
2-6)Hygromycin抗性PDA固体培养基
使用前每1L PDA固体培养基中加入25mg潮霉素。
2-7)YEPD培养基,用于大丽轮枝菌菌丝培养
实施例2构建基因敲除载体
采用Split-marker方法对大丽轮枝菌VdKu80基因敲除载体进行构建。
1、扩增大丽轮枝菌VdKu80基因(VdKu80基因)上、下游片段
1A、根据大丽轮枝菌(Verticillium dahliae Kleb.)野生株XS11基因组VdKu80编码基因的序列,设计2对PCR扩增特异性引物(引物名称、序列如表1所示);其中:Ku80-5F/Ku80-5R为VdKu80基因上游片段引物;Ku80-3F/Ku80-3R为VdKu80基因下游片段引物;
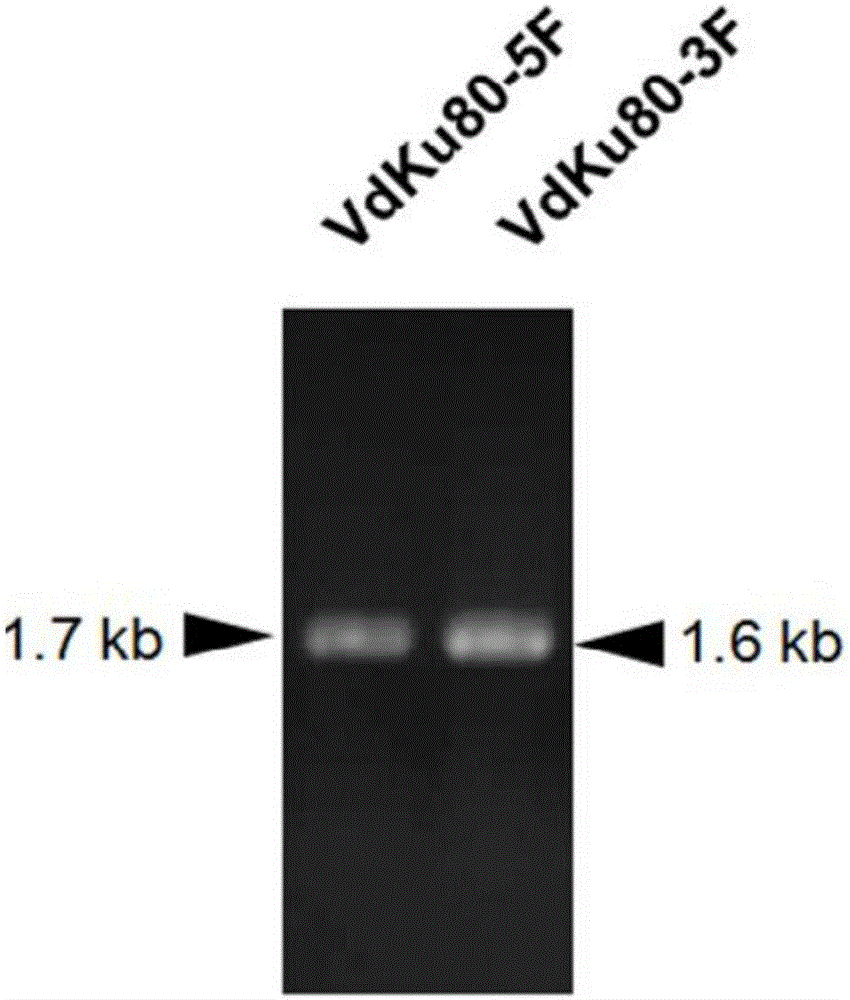
1B、以大丽轮枝菌基因组(完整的基因序列参见http://genome.jgi.doe.gov/Verda1/Verda1.home.html)为模板,分别以Ku80-5F/Ku80-5R;Ku80-3F/Ku80-3R为引物进行PCR扩增,得到1.7kb的VdKu80基因上游片段(VdKu80 5F,SEQ ID NO.36);1.6kb的VdKu80基因下游片段(VdKu80 3F,SEQ ID NO.37);PCR扩增产物的1%琼脂糖凝胶电泳检测结果如图1A所示。
PCR反应体系:
PCR反应条件:94℃预变性5min,94℃30s,55℃30s,72℃2min,30个循环,最后72℃延伸10min,以双蒸水为阴性对照。
2、扩增潮霉素抗性基因片段(Hygromycin片段)
以质粒gGFP为模板,以Hyg-For/Hyg-Rev为引物(引物名称、序列如表1所示),进行PCR扩增,获得1.5kb的Hygromycin片段(即潮霉素抗性基因片段),包括启动子、开放式阅读框和终止子,委托英潍捷基(上海)贸易有限公司进行测序,SEQ ID NO.38。
3、制备重组质粒
3A、将VdKu80 5F、VdKu80 3F片段和Hygromycin片段分别与pMDTM19-T Vector质粒进行连接反应(按照pMDTM19-T Vector clone kit试剂盒说明进行),其中反应体系和条件如下:
目标片段 2μl
pMDTM19-T Vector 0.5μl
Solution I 2.5μl
37℃连接3h
3B、使用DH5α大肠杆菌感受态进行重组质粒的大肠杆菌转化,并分别使用M13F/M13R引物对菌落进行阳性克隆筛选、将能够分别扩增出VdKu80 5F、VdKu80 3F和Hygromycin片段目的条带的菌落进行摇菌,并利用天根生化科技(北京)有限公司的质粒提取试剂盒提取质粒,获得VdKu80 5F重组质粒、VdKu80 3F重组质粒、Hygromycin重组质粒。
4、含有重叠列片段的PCR扩增
以VdKu80 5F重组质粒为模版,以Ku80-5F/Ku80-5RO为引物,进行PCR扩增得到VdKu80 5F重叠扩增片段(即VdKu80 5FO片段);以VdKu80 3F重组质粒为模版,以Ku80-3FO/Ku80-3R为引物,进行PCR扩增得到VdKu80 3F重叠扩增片段(即VdKu80 3FO片段);以Hygromycin重组质粒为模版,以M13F/M13R为引物,进行PCR扩增得到Hygromycin重叠扩增片段(即HygromycinO片段);其中VdKu80 5FO片段在3’端含有一段与M13F相同的DNA序列;VdKu80 3FO片段在5’端含有一段与M13R相同的DNA序列;HygromycinO片段的5’和3’分别含有M13F和M13R的DNA序列;引物序列如表1所示。
PCR反应体系:
PCR反应条件:94℃预变性5min,94℃30s,55℃30s,72℃2min,30个循环,最后72℃延伸10min,以双蒸水为阴性对照。
5、融合PCR
以VdKu80 5FO片段和HygromycinO片段为模板,以Ku80-5F/HY-R为引物(见表1),利用融合PCR方法进行PCR扩增,获得VdKu80基因敲除载体片段1(即VdKu80 5F+2/3Hyg片段);以VdKu80 3FO片段和HygromycinO片段为模板,以YG-F/Ku80-3R为引物(见表1)进行PCR扩增,即通过融合PCR的方法扩增获得VdKu80基因敲除载体片段2(即2/3Hyg+VdKu80 3F片段);融合PCR扩增产物经1%琼脂糖凝胶电泳检测获得的VdKu80敲除载体片段1、2的大小分别为2.7kb、2.5kb。电泳检测结果如图1B。
融合PCR反应体系:
PCR反应条件:94℃预变性5min,94℃30s,55℃45s,72℃3min,30个循环,最后72℃延伸10min,以双蒸水为阴性对照。
实施例3PEG介导的原生质体转化
将VdKu80敲除载体VdKu80 5F+2/3Hyg和2/3Hyg+VdKu80 3F两个片段使用PEG介导的遗传转化方法导入大丽轮枝菌野生型原生质体内。具体转化方法参考(Goswami,RS.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法进行,具体操作如下:
1、将适量的野生型大丽轮枝菌菌株XS11的分生孢子置于液体的YEPD培养基中,于25℃,150rpm摇培一定时间获得新鲜的大丽轮枝菌菌丝
2、利用适量比例的Lyzing和Driselase酶,对大丽轮枝菌新鲜菌丝进行酶解,获得足量的原生质体。
3、将构建好的基因敲除载体VdKu80 5F+2/3Hyg和2/3Hyg+VdKu80 3F片段与大丽轮枝菌原生质体充分混合后,在一定浓度的PEG条件下导入大丽轮枝菌原生质体内。
4、原生质体再生培养
将一定量的TB3液体培养基加入步骤3中的大丽轮枝菌原生质体悬浮液中,于25℃,90rpm进行原生质体再生培养,促进原生质体细胞壁的再生,将细胞壁再生后的菌体置于含25μg/ml潮霉素的TB3固体培养基中生长,共获得13个VdKu80基因敲除的候选转化子。
实施例4基因敲除突变体筛选、鉴定
1、VdKu80基因敲除转化子单胞纯化
将VdKu80基因敲除转化子转接至含潮霉素的PDA固体培养基(即Hygromycin抗性PDA固体培养基,其中潮霉素浓度为25μg/mL)上,于室温下黑暗培养7天后用蒸馏水冲洗菌落,收集转化子的分生孢子,并用10倍梯度稀释法稀释孢子悬液;
将少量的稀释后的孢子悬浮液涂布到新的含Hygromycin的PDA固体培养基上生长,其中潮霉素浓度为25μg/mL,室温下黑暗培养2-3天,待菌落可见,挑取单个菌落于无Hygromycin抗性的PDA平板(即不含潮霉素的PDA固体培养基)上生长,室温下黑暗培养5-7天,用1ml蒸馏水冲洗菌落,收集单胞转化子的分生孢子,并用15%甘油进行保存,置于-80℃待用。经初步筛选,挑取5个单胞转化子(分别为VdKu80-1、VdKu80-4、VdKu80-5、VdKu80-6、VdKu80-9)作为VdKu80基因敲除的待选转化子用于进一步分析验证。
2、突变体的初步筛选
使用CTAB(十六烷基三甲基溴化铵,cetyltrimethylammonium Ammonium Bromide)的方法分别提取每个单胞转化子、野生型菌株(WT)的基因组DNA,使用引物Ku80-IF/Ku80-IR(序列见表1)进行PCR扩增检测。
PCR扩增产物的琼脂糖凝胶电泳检测结果如图2A,野生型菌株和转化子VdKu80-5能够扩增出目的条带(即102bp的VdKu80基因片段),而转化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9未扩增出目的条带。
转化子中VdKu80基因被敲除,则PCR扩增为阴性结果,即PCR扩增产物中不能扩增出102bp大小的目的条带(即VdKu80基因片段),如果仍能扩增出与野生型条带大小(102bp)一样的的产物,则说明VdKu80基因未被敲除。
图2A的检测结果显示:大丽轮枝菌野生型菌株(WT)具有明显的102bp大小的目的条带;转化子VdKu80-5的电泳图中具有与野生型一样大小的条带;而转化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9的PCR扩增产物中未能扩增出102bp大小的条带,因此初步筛选出转化子VdKu80-1、VdKu80-4、VdKu80-6、VdKu80-9为VdKu80基因敲除突变体命名为△VdKu80-1、△VdKu80-4、△VdKu80-6、△VdKu80-9。
3、Southern blot分析、鉴定
使用DIG High Prime DNA Labling and Detection Starter Kit I(Roche)试剂盒,并严格按照其说明进行操作,对VdKu80敲除突变体进行Southern blot分析、鉴定,具体如下:
3A、以VdKu80 3F重组质粒为模板,以Ku80-3F/Ku80-P1为引物(见表1),进行PCR扩增,然后以DIG-High Prime(DIG High Prime DNA Labling and Detection Starter Kit I(Roche)试剂盒)进行标记,获得DIG(地高辛)标记探针;
3B、将大丽轮枝菌野生型菌株、单胞转化子的基因组DNA各约30μg,分别用限制性内切酶KpnI酶(约300U)进行酶切处理;将酶切后的DNA片段经琼脂糖凝胶电泳分析后转移至N+的硝酸纤维素膜上;再利用地高辛(DIG)标记探针进行杂交;然后按照DIG High Prime DNA Labling and Detection Starter Kit I(Roche)试剂盒中的说明对杂交后的硝酸纤维素膜进行洗膜、显色、拍照,结果如图2B所示。
在Southern blot分析、鉴定试验中,野生型经过Southern blot分析杂出一条5.2kb的条带(含有Ku80基因片段);成功敲除VdKu80基因的突变体未检测到该条带,而是杂出一条3.5kb的条带(含有潮霉素基因片段);异位突变体杂出了2条条带,一条与野生型杂出的条带大小一致,另一条与VdKu80敲除突变体杂出的条带大小一致。
由图2A、2B的检测结果可知本发明成功获得4株单拷贝敲除VdKu80基因的大丽轮枝菌突变体菌株,潮霉素抗性基因成功整合并替代了VdKu80基因,命名为VdXS11-△VdKu80-1、VdXS11-△VdKu80-4、VdXS11-△VdKu80-6、VdXS11-△VdKu80-9。本发明将大丽轮枝菌VdKu80敲除突变体菌株VdXS11-△VdKu80-9提交中国微生物菌种保藏管理委员会普通微生物中心进行保藏,其微生物保藏编号为:CGMCC No.12873,保藏于中国微生物菌株保藏管理委员会普通微生物中心;保藏地址:中国北京市朝阳区北辰西路1号院3号中国科学院微生物研究所;保藏日期:2016年8月17日。
实施例5基因敲除突变体表型特征分析
1、VdKu80敲除突变体生长速率、菌落形态、分生孢子产量测定。
1A、生长速率测定:
将新鲜生长的大丽轮枝菌VdKu80基因敲除突变体和野生型大丽轮枝菌菌株的菌丝块(直径0.5cm)分别接种于PDA、CM固体平板培养基中央,室温条件下培养10天后,分别测量菌落直径,测定结果如图3A,作为衡量其生长速率的标准。
测定结果表明:大丽轮枝菌野生型菌株与敲除VdKu80基因的突变体菌株在PDA或者CM固体培养基上菌落直径无显著差异。表明敲除VdKu80基因不影响真菌的生长。
1B、菌落形态观察:
将新鲜生长的大丽轮枝菌VdKu80基因敲除突变体和野生型大丽轮枝菌菌株的菌丝块(直径0.5cm)分别接种于PDA、CM固体平板培养基中央,室温条件下培养,直接拍照观察菌落形态,观察结果如图3B。
VdKu80敲除突变体菌株和野生型菌株不论在PDA培养基上还是在CM培养基上,菌落形态均无显著差异。表明敲除VdKu80基因不影响真菌的菌落形态。
1C、分生孢子产量测定:
将新鲜生长的大丽轮枝菌VdKu80基因敲除突变体和野生型大丽轮枝菌菌株的菌丝块(直径0.5cm)分别接种于PDA固体平板培养基中央,室温条件下培养10天,然后用2ml蒸馏水冲洗菌落,获得分生孢子悬浮液,并用血球计数板对分生孢子数量进行统计,统计结果如图3C。
由图3C可知:本发明的VdKu80敲除突变体菌株和野生型菌株在分生孢子产量方面无显著不同,均产生大量分生孢子,室温条件下培养10天后的分生孢子产量为2.4-2.6×108个/ml,表明敲除大丽轮枝菌的VdKu80基因,并不影响菌株的生长、菌落形态和分生孢子产量。
2、胁迫响应
将新鲜生长的大丽轮枝菌VdKu80基因敲除突变体和野生型大丽轮枝菌菌株的菌丝块(直径0.5cm)分别接种至含有100μg/ml Cong Red(刚果红)、1M/L的NaCl的CM固体平板培养基的中央,室温条件下生长10天,测量大丽轮枝菌野生型和VdKu80敲除突变体菌株的菌落直径,测定结果如图3A所示;观察菌落形态,观察结果如图3B所示。
测定结果表明:野生型菌株和VdKu80基因敲除突变体菌株在胁迫培养基上的菌丝生长较无胁迫的CM固体培养基,生长受到了显著的抑制。尤其是在含1M/L NaCl的CM固体培养基。但是不论是在含100μg/mlCong Red的CM固体培养基还是在含1M/L NaCl的CM固体培养基,野生型菌株和VdKu80基因敲除突变体菌株菌落直径和菌落形态均无显著差异。表明,VdKu80敲除突变体菌株和野生型菌株对细胞壁胁迫剂和渗透压力的抗性一致,敲除VdKu80基因并影响大丽轮枝菌对外界压力的响应。
3、微菌核形成观察
将1×106个孢子/ml的VdKu80敲除突变体和野生型菌株的孢子液均匀的涂布在BM固体培养基上,室温条件下生长5天后,用显微镜观察微菌核形成情况,显微镜观察结果如图4。
VdKu80基因敲除突变体菌株能够形成与野生型菌株类似的黑色素化的微菌核,形成的微菌核无显著差异,敲除VdKu80基因并不影响大丽轮枝菌微菌核的形成。
4、致病性实验
将烟草幼苗的根分别浸泡在分生孢子为1×106个/ml的VdKu80敲除突变体、野生型菌株的孢子悬浮液中10min,然后将烟草重新种植到土壤内,至于温室内生长20-30天后观察烟草幼苗的生长状况以及枯萎病症;对照组(CK)除了采用蒸馏水浸泡烟草幼苗根之外,其余与分生孢子悬浮液浸泡相同。观察结果如图5所示。
经大丽轮枝菌VdKu80基因敲除突变体菌株与野生型菌株浸泡后的烟草幼苗生长状况差,出现了明显的枯萎病症状包括叶片黄萎、脱落;与对照组烟草幼苗相比,接种了丽轮枝菌VdKu80基因敲除突变体菌株与野生型菌株的烟草幼苗生长受到显著的抑制;而且接种大丽轮枝菌VdKu80基因敲除突变体菌株的烟草幼苗与接种大丽轮枝菌野生型菌株的烟草幼苗表型无显著差异。实验表明,敲除大丽轮枝菌VdKu80基因并不影响大丽轮枝菌的致病性。
综上所述,本发明获得的大丽轮枝菌VdKu80敲除突变体菌株在形态上与野生型菌株无显著差异,同时在大丽轮枝菌中敲除VdKu80并不影响菌株的生长、分生孢子的产量、胁迫响应能力、微菌核形成和致病性。本发明获得的VdKu80敲除突变体菌株与野生型菌株在各方面表型上均无显著差异,因此本发明获得的VdKu80敲除突变体菌株可以作为后续基因功能研究的受体菌株,用于基因敲除。
由于VdKu80基因敲除突变体菌株中含有潮霉素抗性筛选基因,因此本实例中使用的是遗传霉素(Geneticin,G418)抗性基因替换VDAG_00736基因、VDAG_07169基因。
试验例1以VdKu80敲除突变体菌株VdXS11-△VdKu80-9作为受体菌株进行VDAG_00736基因敲除效率评估
采用Split-marker方法对大丽轮枝菌VdKu80敲除突变体菌株VdXS11-△VdKu80-9的VDAG_00736基因敲除载体进行构建。
1.1构建VDAG_00736基因敲除载体
按照实施例2构建VdKu80基因敲除载体的方法,构建了VDAG_00736融合遗传霉素抗性的敲除片段VDAG_00736-5F+2/3G418和2/3G418+VDAG_00736-3F。
以大丽轮枝菌基因组DNA(完整的基因序列参见http://genome.jgi.doe.gov/Verda1/Verda1.home.html)为模板,分别以根据大丽轮枝菌VDAG_00736基因的上、下游DNA序列所设计2对PCR扩增特异性引物VDAG_00736-5F/VDAG_00736-5R;VDAG_00736-3F/VDAG_00736-3R为引物进行PCR扩增,获得大丽轮枝菌VDAG_00736基因上游片段VDAG_00736 5F和VDAG_00736基因下游片段VDAG_00736 3F;
以质粒pSilent-Dual1为模板,以根据质粒pSilent-Dual1的DNA序列而设计1对遗传霉素抗性基因PCR扩增的特异性引物Geneticin-For/Geneticin-Rev为引物进行PCR扩增,获得Geneticin片段(即遗传霉素抗性基因片段),包括启动子、开放式阅读框和终止子;
将VDAG_00736 5F、VDAG_00736 3F、Geneticin片段分别与pMDTM19-T Vector质粒进行连接反应,使用DH5α大肠杆菌感受态进行重组质粒的大肠杆菌转化,并分别使用M13F/M13R引物对菌落进行阳性克隆筛选、利用天根生化科技(北京)有限公司的质粒提取试剂盒提取质粒,获得重组质粒VDAG_00736 5F、VDAG_00736 3F、Geneticin;
以VDAG_00736 5F重组质粒为模版,以VDAG_00736-5F/VDAG_00736-5O为引物,进行PCR扩增得到VDAG_00736 5FO片段,该片段在3’端含有一段与M13F引物相同的DNA序列;以VDAG_00736 3F重组质粒为模版,以VDAG_00736-3O/VDAG_00736-3R为引物,进行PCR扩增得到VDAG_00736 3FO片段,该片段在5’端含有一段与M13R引物相同的DNA序列;以Geneticin重组质粒为模版,以M13F/M13R为引物,进行PCR扩增得到5’和3’分别含有M13F和M13R引物序列的GeneticinO片段;
以VDAG_00736 5FO片段和GeneticinO片段为模板,以VDAG_00736-5F/GE-R为引物,利用融合PCR方法进行PCR扩增,获得VDAG_00736基因敲除载体片段A(即VDAG_00736-5F+2/3G418片段);以VDAG_00736 3FO片段和GeneticinO片段为模板,以EN-F/VDAG_00736-3R为引物,通过融合PCR的方法扩增获得VDAG_00736基因敲除载体片段B(即2/3G418+VDAG_00736-3F片段);
1.2PEG介导的原生质体转化
使用PEG介导的遗传转化方法,将构建的VDAG_00736敲除片段VDAG_00736-5F+2/3G418,2/3G418+VDAG_00736-3F导入本发明方法制备的VdXS11-△VdKu80-9敲除突变体菌株的原生质体内,获得VDAG_00736基因敲除的待选转化子,具体转化方法参考(Goswami,R S.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法进行。
1.3VDAG_00736基因敲除突变体筛选和统计
将步骤2中获得的获得VDAG_00736基因敲除的待选转化子转接至含有50μg/ml Geneticin的PDA抗性平板培养基中,于室温条件下黑暗培养7天,用1ml蒸馏水冲洗菌落,收集分生孢子;接着将少量分生孢子悬浮液均匀的涂布到含有50μg/ml Geneticin的PDA抗性平板培养基上进行单胞纯化,挑取单胞转化子;然后将纯化后的转化子保存并采用CTAB法提取基因组DNA;再接着以提取的转化子的DNA为模板,以VDAG_00736-IF/VDAG_00736-IR为引物筛选VDAG_00736基因敲除突变体,并统计敲除突变体的比例(敲除突变体占所有筛选转化子的比例),本试验例中所使用的引物如表1所示,统计结果如表2。
试验例2以VdKu80敲除突变体菌株VdXS11-△VdKu80-9作为受体菌株进行VDAG_07169基因敲除效率评估
采用Split-marker方法对大丽轮枝菌VdKu80敲除突变体菌株VdXS11-△VdKu80-9的VDAG_07169基因敲除载体进行构建。
1、构建VDAG_07169单基因敲除载体
按照实施例2构建VdKu80基因敲除载体的方法,构建了VDAG_07169融合遗传霉素抗性的敲除片段VDAG_07169-5F+2/3G418和2/3G418+VDAG_07169-3F。
以大丽轮枝菌基因组DNA(完整的基因序列参见http://genome.jgi.doe.gov/Verda1/Verda1.home.html)为模板,分别以根据大丽轮枝菌VDAG_07169基因的上、下游DNA序列所设计2对PCR扩增特异性引物VDAG_07169-5F/VDAG_07169-5R、VDAG_07169-3F/VDAG_07169-3R为引物进行PCR扩增,获得大丽轮枝菌VDAG_07169基因上游片段VDAG_07169 5F和VDAG_07169基因下游片段VDAG_07169 3F;
将VDAG_07169 5F和VDAG_07169 3F片段分别与pMDTM19-T Vector质粒进行连接反应,使用DH5α大肠杆菌感受态进行重组质粒的大肠杆菌转化,并分别使用M13F/M13R引物对菌落进行阳性克隆筛选、利用天根生化科技(北京)有限公司的质粒提取试剂盒提取质粒,获得VDAG_07169 5F重组质粒和VDAG_07169 3F重组质粒;
以VDAG_07169 5F重组质粒为模版,以VDAG_07169-5F/VDAG_07169-5O为引物,进行PCR扩增得VDAG_07169 5FO片段,该片段在3’端含有一段与M13F引物相同的DNA序列;以VDAG_07169 3F重组质粒为模版,以VDAG_07169-3O/VDAG_07169-3R为引物,进行PCR得VDAG_07169 3FO片段,该片段在5’端含有一段与M13R引物相同的DNA序列;
以VDAG_07169 5FO片段和试验例1中获得的GeneticinO片段为模板,以VDAG_07169-5F/GE-R为引物,利用融合PCR方法进行PCR扩增,获得VDAG_07169基因敲除载体片段A(即VDAG_07169-5F+2/3G418片段);以VDAG_07169 3FO片段和试验例1中获得的GeneticinO片段为模板,以EN-F/VDAG_07169-3R为引物,通过融合PCR的方法扩增获得VDAG_7169基因敲除载体片段B(即2/3G418+VDAG_07169-3F片段);
2、PEG介导的原生质体转化
使用PEG介导的遗传转化方法,将构建的VDAG_07169敲除片段VDAG_07169-5F+2/3G418,2/3G418+VDAG_07169-3F导入本发明方法制备的VdXS11-△VdKu80-9敲除突变体菌株的原生质体内,获得VDAG_07169基因敲除的待选转化子,具体转化方法参考(Goswami,R S.Targeted gene replacement in fungi using a split-marker approach.Methods Mol Biol,2011,835:255-269)的方法进行。
3、VDAG_07169敲除突变体筛选和统计
将步骤2中获得的VDAG_07169基因敲除的待选转化子转接至含有50μg/ml Geneticin的PDA抗性平板培养基中,于室温条件下黑暗培养7天,用1ml蒸馏水冲洗菌落,收集分生孢子;接着将少量分生孢子悬浮液均匀的涂布到含有50μg/ml Geneticin的PDA抗性平板培养基上进行单胞纯化,挑取单胞转化子;然后将纯化后的转化子分生孢子进行甘油保存并采用CTAB法提取基因组DNA;再接着以提取的转化子DNA为模板,以VDAG_07169-IF/VDAG_07169-IR为引物筛选VDAG_07169基因敲除突变体,并统计敲除突变体的比例(敲除突变体占所有筛选转化子的比例),本试验例中所使用的引物如表1所示;统计结果如表2所示。
表2野生型菌株和△VdKu80突变体菌株基因敲除率的对比
为了验证VdXS11-△VdKu80-9敲除突变体菌株确实具有显著提高基因敲除效率的特性,本发明选取了两个基因VDAG_00736和VDAG_07169作为敲除对象。本发明成功构建了VDAG_00736和VDAG_07169融合Geneticin的敲除片段,经过转化成功获得了与用野生型大丽轮枝菌菌株转化相当的转化子数量。
分别使用VDAG_00736和VDAG_07169基因特异性引物,筛选使用野生型菌株和使用VdXS11-△VdKu80-9敲除突变体菌株获得的具有遗传霉素抗性的转化子。结果表明使用野生型菌株进行转化获得的转化子,均没有筛选得到VDAG_00736和VDAG_07169敲除突变体,敲除效率接近为0。而使用VdXS11-△VdKu80-9敲除突变体菌株进行转化获得转化子,其基因敲除率大约为20%-25%,其中VDAG_00736的敲除效率为25%,VDAG_07169的敲除效率为21.4%(表2)。
研究表明,本发明获得的大丽轮枝菌VdKu80敲除突变体菌株能够显著提高基因敲除率,同时敲除VdKu80对于大丽轮枝菌生长、产孢、微菌核形成、致病性等表型无显著影响。因此本发明获得的大丽轮枝菌VdKu80敲除突变体菌株对于后续的基因功能研究具有巨大的实用价值。
- 还没有人留言评论。精彩留言会获得点赞!