基因组编辑系统及其用途的制作方法
本发明涉及一种基因组编辑系统及其用途,特别涉及一种通过II型启动子驱动的CRISPR/Cas9系统及其用途。
背景技术:
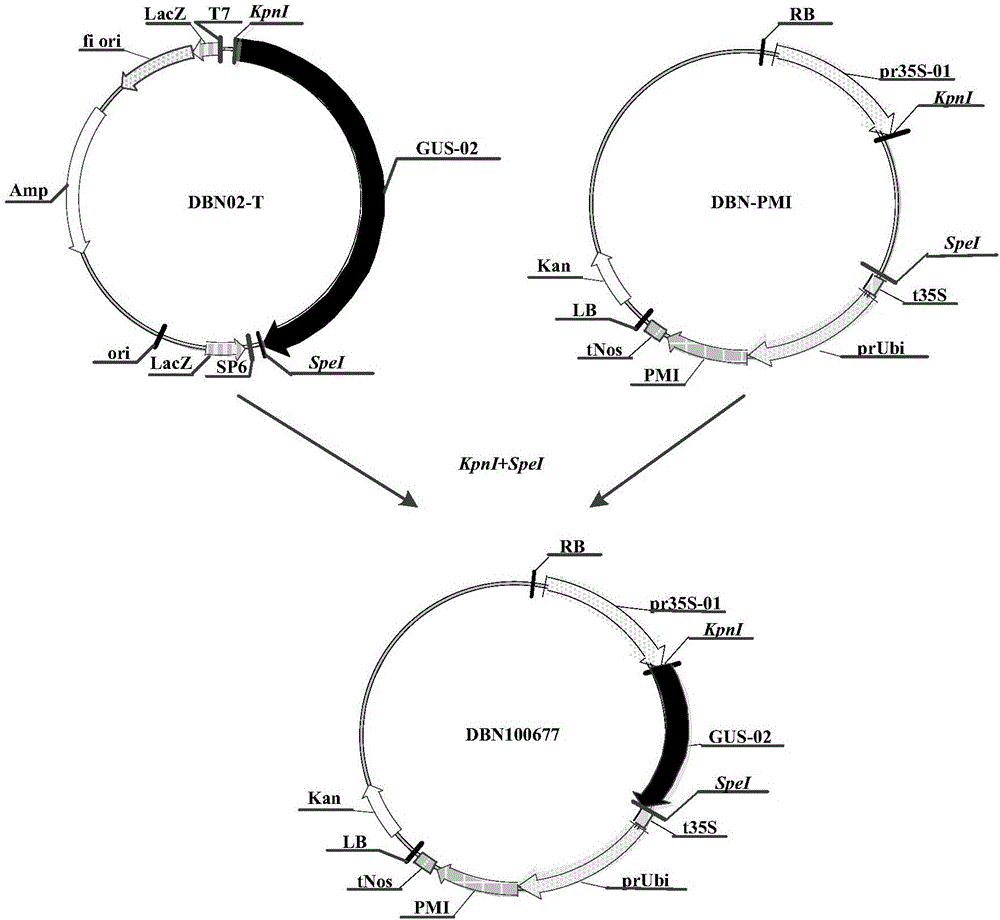
:CRISPR是一种来自细菌降解入侵的病毒DNA或其他外源DNA的免疫机制。在细菌及古细菌中,CRISPR系统共分成3类,其中Ⅰ类和Ⅲ类需要多种CRISPR相关蛋白(Cas蛋白)共同发挥作用,而Ⅱ类系统只需要一种Cas蛋白即可,这为其能够广泛应用提供了便利条件。涉及Cas9蛋白和sgRNA的Ⅱ型CRISPR/Cas系统是代表性的,在sgRNA的引导下,Cas9蛋白首先与sgRNA结合成复合物,然后通过PAM序列结合并侵入DNA,形成RNA-DNA复合结构,进而对目的DNA双链进行切割,使DNA双链断裂,利用细胞自身的非同源性末端接合和同源重组修复机制实现错误修复或者引入改变,造成DNA序列改变,从而实现基因的定向修饰操作。在植物中,通常使用的CRISPR/Cas系统包含sgRNA和Cas9两个独立的表达盒,并分别由III型启动子(如水稻U3或U6启动子等)和II型启动子(如花椰菜花叶病毒35S启动子或泛素蛋白1启动子等)驱动表达。该系统主要存在如下缺陷:(1)sgRNA一般由水稻U3或U6启动子驱动表达,但二者要求转录的第一个碱基分别是A和G,因此其对靶点的选择具有一定的限制。(2)水稻U3和U6启动子均为组成型启动子,无法实现在特异组织中对不同的基因进行编辑。(3)如果Cas9表达盒的启动子与选择性标记(如Hpt或PMI等)表达盒的启动子相同,在某些品种的农杆菌中便容易发生因重组而导致的Cas9元件丢失的现象,导致农杆菌转化后获得的转基因植株基本上没有突变体,进而影响CRISPR/Cas9系统的应用效果。技术实现要素:本发明的目的是提供一种基因组编辑系统及其用途,有效克服了现有技术中III型启动子对转录起始位点的碱基限制、不能在特异组织中对不同的基因进行编辑、由于重组而导致元件丢失导致无法获得突变体等技术缺陷。为实现上述目的,本发明提供一种嵌合序列,包含sgRNA序列和可剪切序列,所述sgRNA序列插入间插序列的内部。进一步地,所述sgRNA序列插入所述间插序列内部的位置两侧各至少保留长度为12bp的所述间插序列。在上述技术方案的基础上,所述嵌合序列还包括靶点序列。具体地,所述间插序列为内含子序列。为实现上述目的,本发明还提供一种基因组编辑系统,包括a)和b):a)所述嵌合序列;b)Cas9蛋白。进一步地,所述嵌合序列与编码所述Cas9蛋白的核酸序列有效连接。所述嵌合序列有效连接于调控序列。在上述技术方案的基础上,所述调控序列包括II型启动子和/或定位结构域。优选地,所述II型启动子包括组成型启动子、组织特异性启动子和诱导型启动子。具体地,所述组成型启动子包括花椰菜花叶病毒35S启动子或泛素启动子。所述组织特异性启动子包括花粉特异性表达启动子、根特异性表达启动子、绿色组织特异性表达启动子或胚乳特异性表达启动子。所述诱导型启动子包括化学诱导型启动子、冷诱导型启动子、热诱导型启动子或病毒诱导型启动子。为实现上述目的,本发明还提供一种由II型启动子驱动的基因组编辑系统,包括a)、b)和c):a)II型启动子;b)所述嵌合序列;c)Cas9蛋白。进一步地,所述嵌合序列与编码所述Cas9蛋白的核酸序列有效连接。所述嵌合序列有效连接于所述II型启动子。优选地,所述II型启动子包括组成型启动子、组织特异性启动子和诱导型启动子。具体地,所述组成型启动子包括花椰菜花叶病毒35S启动子或泛素启动子。所述组织特异性启动子包括花粉特异性表达启动子、根特异性表达启动子、绿色组织特异性表达启动子或胚乳特异性表达启动子。所述诱导型启动子包括化学诱导型启动子、冷诱导型启动子、热诱导型启动子或病毒诱导型启动子。为实现上述目的,本发明还提供一种表达盒,包含编码所述基因组编辑系统的核酸序列或编码所述由II型启动子驱动的基因组编辑系统的核酸序列。为实现上述目的,本发明还提供一种重组表达载体,包含编码所述基因组编辑系统的核酸序列或或编码所述由II型启动子驱动的基因组编辑系统的核酸序列或所述表达盒。为实现上述目的,本发明还提供一种实现基因组编辑的方法,包括在生物体中表达所述基因组编辑系统或所述由II型启动子驱动的基因组编辑系统。为实现上述目的,本发明还提供一种编辑靶位点序列的方法,包括将所述基因组编辑系统导入包含靶位点序列的生物体中。为实现上述目的,本发明还提供一种产生基因组编辑的植物的方法,包括向植物基因组中引入编码所述基因组编辑系统的核酸序列或编码所述由II型启动子驱动的基因组编辑系统的核酸序列。为实现上述目的,本发明还提供一种产生基因组编辑植物种子的方法,包括将所述方法产生的基因组编辑的植物自交,从而获得具有基因组编辑植物种子。为实现上述目的,本发明还提供一种培育基因组编辑植物的方法,包括:种植至少一粒所述方法产生的所述基因组编辑植物种子;使所述种子长成植株。为实现上述目的,本发明还提供一种所述基因组编辑系统或所述由II型启动子驱动的基因组编辑系统在获得基因组突变生物中的用途。为实现上述目的,本发明还提供一种试剂盒,包括所述基因组编辑系统或所述由II型启动子驱动的基因组编辑系统,用于实现基因组编辑。本发明中所述的CRISPR(ClusteredRegularlyInterspacedShortPalindromicRepeats),是指成簇的规律间隔的短回文重复序列,含有多个短同向重复的基因座,其被发现存在于约40%测序细菌的基因组中和90%测序古细菌的基因组中。CRISPR作为原核的免疫系统发挥功能,其赋予对外来遗传元件例如质粒和噬菌体的抵抗性。CRISPR系统提供了一种获得性免疫形式。外源DNA的短片段(称为间隔区)整合在CRISPR重复序列之间的基因组中,然后CRISPR间隔区以类似于真核生物中RNAi的方式用于识别和沉默外来遗传元件。在细菌及古细菌中,CRISPR系统共分成3类,其中Ⅰ类和Ⅲ类需要多种CRISPR相关蛋白(Cas蛋白)共同发挥作用,而Ⅱ类系统只需要一种Cas蛋白即可,这为其能够广泛应用提供了便利条件。本发明所述Cas蛋白的非限制性实例包括:Cas1、Cas1B、Cas2、Cas3、Cas4、Cas5、Cas6、Cas7、Cas8,Cas9(也称为Csn1和Csx12)、Cas10、Csy1、Csy2、Csy3、Cse1、Cse2、Csc1、Csc2、Csa5、Csn2、Csm2、Csm3、Csm4、Csm5、Csm6、Cmr1、Cmr3、Cmr4、Cmr5、Cmr6、Csb1、Csb2、Csb3、Csx17、Csx14、Csx10、Csx16、CsaX、Csx3、Csx1、Csx15、Csf1、Csf2、Csf3、Csf4、其同系物、或其修饰形式。这些酶是已知的;例如,化脓链球菌Cas9蛋白的氨基酸序列可见于SwissProt数据库登录号Q99ZW2下。在一些实施例中,未修饰的CRISPR酶如Cas9具有DNA切割活性。在一些实施例中,该CRISPR酶是Cas9,并且可以是来自化脓链球菌或肺炎链球菌的Cas9。本发明中所述的Cas9,Ⅱ型CRISPR/Cas系统中一种重要的蛋白质成分,可以从生物体如链球菌属物种(Streptococcussp.),优选酿脓链球菌(Streptococcuspyogenes)中分离得到。当Cas9与称为CRISPRRNA(crRNA)和反式激活crRNA(TracrRNA)的两个RNA复合时,形成活性核酸内切酶,从而切断入侵噬菌体或质粒中的外源遗传元件,以保护宿主细胞。crRNA从宿主基因组中的CRISPR元件转录,其中该CRISPR元件之前自外源入侵物捕获。研究表明通过融合crRNA和tracrRNA的必要部分产生的单链嵌合RNA可以取代Cas9/RNA复合体中的两个RNA以形成功能性核酸内切酶。Cas9蛋白质的变体可以是Cas9的突变体形式,其中催化性天冬氨酸残基改变为任何其它氨基酸。优选地,所述其它氨基酸可以是丙氨酸。本发明中所述的向导RNA或引导RNA(guideRNA,gRNA),也称为小向导RNA(smallguideRNA,sgRNA),作用于动质体(kinetoplastid)体内一种称为RNA编辑(RNAediting)的后转录修饰过程中,也是一种小型非编码RNA。可与pre-mRNA配对,并在其中插入一些尿嘧啶(U),产生具有作用的mRNA。向导RNA编辑的RNA分子,长度大约是60-80个核苷酸,是由单独的基因转录的,在gRNA的5’末端有一段锚定区,以特殊的G-U配对方式与非编辑的pre-mRNA序列互补,锚定序列促进gRNA与pre-mRNA中的编辑区互补,特异结合;在gRNA分子的中间部位有一个编辑区负责在被编辑的pre-mRNA分子中插入U的位置,其与被编辑mRNA精确互补;在gRNA分子的3’末端,有一段转录后加入的由大约15个非编码的PolyU序列,功能为把gRNA链接到pre-mRNA的编辑区的5’上游富含嘌呤碱基的核苷酸序列上。在编辑时,形成一个编辑体(editosome),以gRNA内部的序列作为模板进行转录物的校正,同时产生编辑的mRNA。有三种类型CRISPR/Cas系统,其中涉及Cas9蛋白质和crRNA、tracrRNA的Ⅱ型CRISPR/Cas系统是代表性的。Cas9蛋白质通过人工修饰的向导RNA的导向作用可以打靶DNA序列的5’-N20-NGG-3’(N代表任何脱氧核苷酸碱基),N20是与gRNA的5’序列相同的20个碱基,NGG是PAM区(原型间隔子邻近基序,Protospacer-adjacentmotif)。Cas9剪切的位点就是PAM附近的区域。相对于锌指和转录激活因子样效应物DNA结合蛋白提供了优势-因为在核苷酸结合CRISPR-Cas蛋白中的位点特异性由RNA分子调控而不是DNA结合蛋白调控。本发明中所述重组,当用于例如细胞、核酸、蛋白质或载体时,表示该细胞、核酸、蛋白质或载体已通过引入异源核酸或蛋白质、或改变天然核酸或蛋白质而被修饰,或该细胞源自此修饰的细胞。本发明中向导RNA可以以编码该向导RNA的RNA或DNA的形式转移到细胞或生物体中。向导RNA可以是分离的RNA、并入病毒载体的RNA的形式、或者在载体中编码。优选地,载体可以是病毒载体、质粒载体、或农杆菌载体。编码向导RNA的DNA可以是包含编码向导RNA序列的载体。例如,可以通过用分离的向导RNA或包含编码向导RNA的序列和启动子的质粒DNA转染细胞或生物体,将向导RNA转染到细胞或生物体。本发明中所述切割或剪切是指核苷酸分子共价骨架的断裂。向导RNA可以制备为特异于任何待切割的靶,通过向导RNA的靶特异性部分切割任何靶DNA。本发明中所述的植物、植物组织或植物细胞的基因组,是指植物、植物组织或植物细胞内的任何遗传物质,且包括细胞核和质体和线粒体基因组。本发明中所述的多核苷酸和/或核苷酸形成完整“基因”,在所需宿主细胞中编码蛋白质或多肽。本领域技术人员很容易认识到,可以将本发明的多核苷酸和/或核苷酸置于目的宿主中的调控序列控制下。如本申请,包括权利要求中使用的,除非上下文中清楚地另有说明,否则单数和单数形式的术语,例如“一”、“一个”和“该”,包括复数指代物。因此,例如“植物”、“该植物”或“一个植物”也指示多个植物。并且根据上下文,使用术语“植物”还可指示该植物在遗传上相似或相同的后代。类似地,术语“核酸”可以指核酸分子的许多拷贝。类似地,术语“探针”可以指代相同或相似的探针分子。数字范围包括限定该范围的数字,并明确地包括所限定的范围内的每个整数和非整数分数。除非另外指出,否则本文所用的全部技术和科学术语具有与本领域普通技术人员普遍理解的相同的含义。本发明中,术语“核酸”、“核苷酸”、“核苷酸序列”、“寡核苷酸”和“多核苷酸”可互换使用,它们是指具有任何长度的核苷酸的聚合形式,根据上下文含义,可指代DNA或RNA、或其类似物。其中DNA包括但不限于cDNA、基因组DNA、合成DNA(例如人工合成)和含核酸类似物的DNA(或RNA)。多核苷酸可具有任何三维结构,并且可以执行已知或未知的任何功能。核酸可为双链或单链(既有义链或反义单链)。多核苷酸的非限制性示例包括基因、基因片段、外显子、内含子、信使RNA(mRNA)、转移RNA、核糖体RNA、核酶、cDNA、重组多核苷酸、分支多核苷酸、质粒、载体、任何序列的分离DNA、任何序列的分离RNA、核酸探针和引物、以及核酸类似物。本发明中,术语“间插序列”是指基因间或基因内的非编码序列。大多数真核结构基因中的间插序列或不编码序列可以转录,但在基因转录后,由这些间插序列转录的部分经加工被从初级转录本中准确除去,才产生有功能的RNA。本发明中,术语“内含子(Intron)”是真核生物细胞DNA中的间插序列,是阻断基因线性表达的序列。这些序列被转录在前体RNA中,但RNA上的内含子会在RNA离开细胞核进行转译前被剪除,最终不存在于成熟RNA分子中。在成熟mRNA被保留下来的基因部分被称为外显子。内含子有时也叫内显子,与外显子相对。内含子和外显子的交替排列构成了割裂基因。在前体RNA中的内含子常被称作“间插序列”。在转录后的加工中,它比外显子有更多的突变。“内含子”也指编码相应RNA内含子的DNA中的区域。真核基因所含内含子的数目、位置和长度不尽相同。内含子是一段特殊的DNA序列,其可以实现自剪切。本发明使用的内含子来源于蓖麻(Ricinuscommunis)。在植物中,内含子可以稳定和增加基因的表达量。来源于细菌的GUS基因是没有内含子的,故在一般情况下,当在植物中表达GUS时,会在该GUS基因序列内部加入来源于蓖麻的内含子(如pCambia系列的载体),以稳定和增加在植物中的表达。具有内含子功能并在严格条件下与本发明内含子序列或其片段杂交的分离序列包括在本发明中。这些序列与本发明序列至少大约40%-50%同源,大约60%、65%或70%同源,甚至至少大约75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更多同源。即序列同一性的范围分布在至少大约40%-50%、大约60%、65%或70%同源,甚至至少大约75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或更大的序列同一性。本发明中,“嵌合序列”是指利用基因重组技术将来源不同的DNA/RNA序列重组而成的人工序列。本发明中“野生型”表示生物、菌株、基因的典型形式或者当它在自然界存在时区别于突变体或变体形式的特征。本发明中“突变体”或“变体”是指发生突变的个体,其具有与野生型不同的序列,可能会导致其中序列的至少部分功能已丢失的序列,例如,在启动子或增强子区域中序列的变化将至少部分地影响生物体中编码序列的表达。术语“突变”是指可由诸如缺失、添加、取代或重排引起的核酸序列中序列的任何变化。突变还可影响该序列参与的一个或多个步骤。例如,DNA序列中的变化可导致有活性的、有部分活性的或无活性的改变的mRNA和/或蛋白的合成。本发明中“非天然存在的”表明人工的参与。当指核酸分子或多肽时,表示该核酸分子或多肽至少基本上从它们在自然界中或如发现于自然界中的与其结合的至少另一种组分游离出来。本发明中“表达”指感兴趣的序列转录产生对应mRNA并且该mRNA翻译产生对应产物,即肽、多肽或蛋白。调节元件控制或调整感兴趣的序列表达,所述调节元件包括5’调节元件如启动子。本发明中“多肽”、“肽”和“蛋白质”可互换地使用,是指具有任何长度的氨基酸聚合物。该聚合物可以是直链或支链的,它可以包含修饰的氨基酸,并且它可以被非氨基酸中断。这些术语还涵盖已经被修饰的氨基酸聚合物;这些修饰例如二硫键形成、糖基化、脂化、乙酰化、磷酸化、或任何其他操作,如与标记组分的结合。术语“氨基酸”包括天然的和/或非天然的或者合成的氨基酸,包括甘氨酸以及D和L旋光异构体、以及氨基酸类似物和肽模拟物。本发明中术语“载体”是指能够在宿主细胞内复制的DNA分子。质粒和粘粒是示例性载体。此外,术语“载体”和“媒介”可互换使用以指将DNA片段从一种细胞转移至另一种细胞的核酸分子,因此细胞不必要属于相同的生物(例如将DNA片段从农杆菌细胞转移至植物细胞)。本发明中术语“表达载体”是指含有所需要的编码序列和在特定宿主生物中表达有效连接的编码序列所需要的适宜核酸序列的重组DNA分子。本发明中术语“重组表达载体”指来自任何来源、能够整合入基因组或自主复制的任何因子如质粒、粘粒、病毒、BAC(细菌人工染色体)、自主复制型序列、噬菌体、或者线性或环状单链或双链DNA或RNA核苷酸序列,包括其中一种或多种DNA序列使用众所周知的重组DNA技术以功能性可操作方式连接的DNA分子。本发明中所述调控序列包括但不限于启动子、转运肽、终止子、增强子、前导序列以及其它可操作地连接到所述嵌合序列的调节序列。在本发明中,“定位结构域”可任选地添加为蛋白部分的部分,定位结构域可将蛋白部分或编程的蛋白部分或组装的复合物定位至活细胞中特定细胞或亚细胞定位。定位结构域可通过将蛋白部分的氨基酸序列融合到掺入包括以下结构域的氨基酸来构建:核定位信号(NLS);线粒体前导序列(MLS);叶绿体前导序列;和/或被设计以将蛋白运输或引导或定位至含有核酸的细胞器、细胞区室或细胞的任何细分部分的任何序列。在一些实施方案中,生物体是真核生物,且定位结构域包括允许蛋白进入细胞核和基因组DNA内的核定位结构域(NLS)。所述NLS的序列可包括带正电荷序列的任何功能NLS。在另一些实施方案中,定位结构域可包括使蛋白部分或编程的核蛋白进入细胞器的前导序列,使细胞器DNA被修饰成为可能。本发明中,真核生物有3类RNA聚合酶,负责转录3类不同的启动子。由RNA聚合酶I负责转录的rRNA基因,启动子(I型)比较单一,由转录起始位点附近的两部分序列构成:第一部分是核心启动子(corepromoter),由-45—+20位核苷酸组成,单独存在时就足以起始转录;另一部分由-170—-107位序列组成,称为上游调控元件,能有效地增强转录效率。由RNA聚合酶Ⅲ负责转录的是5SrRNA、tRNA和某些核内小分子RNA(snRNA),其启动子(Ⅲ型)组成较复杂,又可被分为两个亚类:一类属于结构基因内启动子,一类属于结构基因外启动子。内启动子的有效启动依赖于基因内部包含的两个不连续的DNA片段,这两个DNA片段包括一些不同的连续DNA序列A、B或C区,两个区之间被隔开。根据两个区的不同组合,可以分为I类和II类两种:I类包括A和C区,目前发现其仅存在于5SrRNA的基因中;II类包括A和B区,存在于tRNA的基因、7SLRNA的基因、腺病毒VAI与VAII的RNA中。A、B或A、C内部DNA序列是转录因子TFⅢA和TFⅢC转录启动结合部位。在多数情况下,5’端还会有其它调节或关键元件,这些元件对RNA的高效转录是必需的。这些序列的存在与否之间影响着转录效率。这些序列呈现复杂的多样性,不过大多数启动子5’端-30到-20处存在着TATA盒样序列,与外启动子相似。外启动子缺乏相应的内部序列,仅在5’端有顺式作用元件,在基因的末端有一组由4个或更多胸腺嘧啶组成的终止信号,如脊椎动物U6核小RNA和7SKRNA启动子,这些启动子都高度相似或完全相同,其位置和碱基序列高度保守,其结构与polII启动子也有一定的相似性。它们的5’端顺式作用元件包括几个控制元件,在其上游约-30处存在一个TATA样序列,近-60处有一个snRNAPSE(snRNA近似序列)和一个或多个被称为OCT的修饰序列5’-ATGCAAAT-3’。TATA样序列是polIII对snRNA基因进行转录特异的。TATA样元件和PSE元件共同决定了转录起始位点的选择和转录效率。TATA样元件和PSE元件之间的距离决定了RNA聚合酶转录的特异性,但似乎TATA样元件更重要,因为在PSE缺失的U6RNA和7SKRNA基因转录中,只有转录效率的下降。而且,PSE元件可能和B盒(boxB)相关,B盒在一定程度上可以替代PSE元件。这些序列对下游基因的转录具有决定性的作用,而且它们与起始位点相距较远,往往超过150bp,而对polIII内启动子来说,一般在80bp以内;与polIII外启动子相比,polII启动子5’端各顺式作用元件的作用刚好相反,如果TATA样元件缺失,PSE则可以履行TATA样元件功能,决定转录的起始部位。在PSE上游,外启动子还有一个远端控制序列,其结构与polII增强子OCT骨架相似,但较其复杂。而在-223处还有一个CACC序列与OCT骨架相连。这些远端控制序列的存在可大大提高U6RNA和7SKRNA的表达效率。由RNA聚合酶II负责转录的II型基因包括所有蛋白质编码基因和部分snRNA基因,后者的启动子结构与III型基因启动子中的第三亚类相似,编码蛋白质的II型基因启动子在结构上有共同的保守序列。转录起始位点没有广泛的序列同源性,但第一个碱基为腺嘌呤,而两侧是嘧啶碱基。这个区域被称为起始子(initiator,Inr),序列可表示为Py2CAPy5。Inr元件位于-3—+5。仅由Inr元件组成的启动子是具有可被RNA聚合酶II识别的最简单启动子形式。多数II型启动子有一个被称为TATA盒的共有序列,通常处于-30区,相对于转录起始位点的位置比较固定。TATA盒存在于所有真核生物中,TATA盒是一个保守的七碱基对,也有一些II型启动子不含有TATA盒,这样的启动子称为无TATA盒启动子。本发明中术语“启动子”是指DNA调节区。本发明所述“组成型启动子”是指在该类启动子的控制下,目的异源核苷酸序列的表达大体恒定在一定水平上,在植物的不同组织和/或不同生长发育阶段表达水平没有明显差异。所述组织为植物体中由来源相同和执行同一功能的一种或多种类型细胞集合而成的结构单位,例如保护组织、输导组织、营养组织、机械组织、分生组织,几种不同的组织有机配合、紧密联系,形成不同的器官(organ),不同的器官之间互相配合,更有效地完成有机体的整个生命活动过程。所述生长发育阶段可根据植物形态、机能的差异划分为胚胎阶段、幼苗阶段、成熟阶段和衰老阶段。本发明中术语“组成型的表达”是指目的异源核苷酸序列在植物的不同组织和/或不同生长发育阶段都以大体上一致的水平进行表达。指导植物内组成型表达的启动子的示例包括但不限于,来源于花椰菜花叶病毒的35S启动子、玉米ubi启动子、水稻GOS2基因的启动子等。可获得的植物相容性启动子的范围包括组织特异性启动子和诱导型启动子。诱导型调控元件是能应答于诱导剂直接或间接激活一条或多条DNA序列或基因的转录的元件。不存在诱导剂时,DNA序列或基因将不能被转录。典型地,与诱导型调控元件特异结合以激活转录的蛋白质因子以无活性形式存在,其再通过诱导剂直接或间接转化为活性形式。诱导剂可以是化学试剂,例如蛋白质、代谢产物、生长调控因子、除草剂或酚类化合物或直接通过热、冷、盐或毒性元素施加或通过病原体肌动蛋白或疾病剂(例如病毒)间接施加的生理胁迫。含有诱导型调控元件的植物细胞可暴露于诱导剂,通过喷雾、浇水、加热或类似方法将诱导剂应用到细胞或植物上。任何诱导型启动子均可用于本发明。示例性的诱导型启动子包括蜕皮素受体启动子;来自ACE1系统的应答于铜的启动子;来自玉米的In2-1和In2-2基因,其应答于苯磺酰胺除草剂安全剂;玉米GST启动子,其由用作为前紧急除草剂的疏水亲电化合物激活;和烟草PR-1a启动子,其由水杨酸激活。其它受化学调控的启动子包括甾醇应答型启动子以及四环素诱导型和四环素遏制型启动子。本发明所述“组织特异性启动子”又称为器官特异性启动子,可用于靶向特定的植物组织中增强的转录和/或表达。启动子可在目标组织中表达也在其它植物组织中表达,可在目标组织中强烈表达以及比其它组织程度低得多的表达,或者可高度优选在目标组织中表达。根据启动子调控的基因表达组织的不同,可分为根特异性启动子、花特异性启动子、胚特异性启动子等。Koehorst-vanPutten等分析GBSSI启动子,发现该启动子可以驱动基因在木薯块状根中的表达,但不能在其余组织中表达,表明该启动子具有组织特异性表达的特性。吕山花等利用GUS稳定表达系统分析了pyk10启动子在转基因烟草中的表达情况,发现该启动子驱动的基因在根中特异性表达。房孝良等研究了水稻OsESP1启动子,发现该启动子驱动的GUS基因只在胚中表达,说明该启动子为胚特异性启动子。还如在绿色组织中指导编码序列的表达水平高于植物的其他组织,如PEP羧化酶启动子。本发明中所述“有效连接”或“可操作地连接”表示核酸序列的联结,所述联结使得一条序列可提供对相连序列来说需要的功能。在本发明中所述“有效连接”可以为将启动子与感兴趣的序列相连,使得该感兴趣的序列的转录受到该启动子控制和调控。当感兴趣的序列编码蛋白并且想要获得该蛋白的表达时“有效连接”表示:启动子与所述序列相连,相连的方式使得得到的转录物高效翻译。如果启动子与编码序列的连接是转录物融合并且想要实现编码的蛋白的表达时,制造这样的连接,使得得到的转录物中第一翻译起始密码子是编码序列的起始密码子。备选地,如果启动子与编码序列的连接是翻译融合并且想要实现编码的蛋白的表达时,制造这样的连接,使得5’非翻译序列中含有的第一翻译起始密码子与启动子相连结,并且连接方式使得得到的翻译产物与编码想要的蛋白的翻译开放读码框的关系是符合读码框的。可以“有效连接”的核酸序列包括但不限于:提供基因表达功能的序列(即基因表达元件,例如启动子、5’非翻译区域、内含子、蛋白编码区域、3’非翻译区域、聚腺苷化位点和/或转录终止子)、提供DNA转移和/或整合功能的序列(即T-DNA边界序列、位点特异性重组酶识别位点、整合酶识别位点)、提供选择性功能的序列(即抗生素抗性标记物、生物合成基因)、提供可计分标记物功能的序列、体外或体内协助序列操作的序列(即多接头序列、位点特异性重组序列)和提供复制功能的序列(即细菌的复制起点、自主复制序列、着丝粒序列)。本发明中,所述调控序列包括但不限于启动子、转运肽、终止子,增强子,前导序列,内含子以及其它可操作地连接到CRISPR系统的一个或多个元件,从而驱动该CRISPR系统的所述一个或多个元件的表达。一般而言,CRISPR(规律间隔成簇短回文重复),也称为SPIDR(Spacer间隔开的同向重复),构成通常对于特定细菌物种而言特异性的DNA基因座的家族。该CRISPR座位包含在大肠杆菌中被识别的间隔开的短序列重复(SSR)的一个不同类、以及相关基因。类似的间隔开的SSR已经鉴定于地中海富盐菌、化脓链球菌、鱼腥草属和结核分枝杆菌中。这些CRISPR座位典型地不同于其它SSR的重复结构,这些重复已被称为规律间隔的短重复(SRSR)。一般而言,这些重复是以簇存在的短元件,其被具有基本上恒定长度的独特间插序列规律地间隔开。虽然重复序列在菌株之间是高度保守的,许多间隔开的重复和这些间隔区的序列一般在菌株与菌株之间不同,已经在40种以上的原核生物中鉴定出CRISPR座位。本发明中,“靶点”、“靶位点”或“靶点序列”、“靶位点序列”是指被作用的任何期望的预定的核酸序列,包括但不限于编码或非编码序列、基因、外显子或内含子、调节序列、基因间序列、合成序列和细胞内寄生物序列。在一些实施方案中,靶点序列存在于靶细胞、组织、器官或生物体内。靶点序列包括靶位点,包括靶点序列内的一个或多个核苷酸,其可通过本发明公开的内容被修饰至任何程度。例如,靶点序列可包含一个核苷酸。例如,靶点序列可包含1-300个核苷酸。例如,靶点序列可包含约1-100个核苷酸。例如,靶点序列可包含1-50个核苷酸。例如,靶点序列可包含约1-35个核苷酸。在一些实施方案中,靶点序列可包括不止一个靶位点,其可以是相同的或不同的。引物的长度一般是11个多核苷酸或更多,优选的是18个多核苷酸或更多,更优选的是24个多核苷酸或更多,最优选的是30个多核苷酸或更多。这种引物在高度严格杂交条件下与目标序列特异性地杂交。尽管不同于目标DNA序列且对目标DNA序列保持杂交能力的引物是可以通过常规方法设计出来的,但是,优选的,本发明中的引物与目标序列的连续核酸具有完全的DNA序列同一性。本发明的引物在严格条件下与目标DNA序列杂交。核酸分子或其片段在一定情况下能够与其他核酸分子进行特异性杂交。如本发明使用的,如果两个核酸分子能形成反平行的双链核酸结构,就可以说这两个核酸分子彼此间能够进行特异性杂交。如果两个核酸分子显示出完全的互补性,则称其中一个核酸分子是另一个核酸分子的“互补物”。如本发明使用的,当一个核酸分子的每一个核苷酸都与另一个核酸分子的对应核苷酸互补时,则称这两个核酸分子显示出“完全互补性”。如果两个核酸分子能够以足够的稳定性相互杂交从而使它们在至少常规的“低度严格”条件下退火且彼此结合,则称这两个核酸分子为“最低程度互补”。类似地,如果两个核酸分子能够以足够的稳定性相互杂交从而使它们在常规的“高度严格”条件下退火且彼此结合,则称这两个核酸分子具有“互补性”。从完全互补性中偏离是可以允许的,只要这种偏离不完全阻止两个分子形成双链结构。为了使一个核酸分子能够作为引物或探针,仅需保证其在序列上具有充分的互补性,以使得在所采用的特定溶剂和盐浓度下能形成稳定的双链结构。如本发明使用的,基本同源的序列是一段核酸分子,该核酸分子在高度严格条件下能够和相匹配的另一段核酸分子的互补链发生特异性杂交。促进DNA杂交的适合的严格条件,例如,大约在45℃条件下用6.0×氯化钠/柠檬酸钠(SSC)处理,然后在50℃条件下用2.0×SSC洗涤,这些条件对本领域技术人员是公知的。例如,在洗涤步骤中的盐浓度可以选自低度严格条件的约2.0×SSC、50℃到高度严格条件的约0.2×SSC、50℃。此外,洗涤步骤中的温度条件可以从低度严格条件的室温约22℃,升高到高度严格条件的约65℃。温度条件和盐浓度可以都发生改变,也可以其中一个保持不变而另一个变量发生改变。优选地,本发明的一个核酸分子可以在中度严格条件下,例如在约2.0×SSC和约65℃下与SEQIDNO:1和SEQIDNO:2中一个或多个核酸分子或其互补序列,或者上述序列的任一片段发生特异性杂交。更优选地,本发明的一个核酸分子在高度严格条件下与SEQIDNO:1和SEQIDNO:2中一个或多个核酸分子或其互补序列,或者上述序列的任一片段发生特异性杂交。本发明中,优选的标记物核酸分子具有SEQIDNO:1或SEQIDNO:2或其互补序列,或者上述序列的任一片段。本发明另一优选的标记物核酸分子与SEQIDNO:1或SEQIDNO:2或其互补序列,或者上述序列的任一片段具有80%到100%或90%到100%的序列同一性。SEQIDNO:1或SEQIDNO:2可以用作植物育种方法中的标记物以鉴定遗传杂交的后代。探针与目标DNA分子的杂交可以通过任何一种为本领域技术人员所熟知的方法进行检测,这些方法包括但不限于,荧光标记、放射性标记、抗体类标记和化学发光标记。关于使用特定的扩增引物对目标核酸序列进行的扩增(例如,通过PCR),“严格条件”指的是在DNA热扩增反应中仅允许引物对目标核酸序列发生杂交的条件,具有与目标核酸序列相应的野生型序列(或其互补序列)的引物,能够与所述目标核酸序列结合,并且优选产生唯一的扩增产物,扩增产物即扩增子。术语“特异性结合(目标序列)”是指在严格杂交条件下引物仅与包含目标序列的样品中的目标序列发生杂交。表达盒可另外含有可选择的标记基因。一般地,表达盒将包含用于选择转化细胞的选择性标记基因。所述选择性标记基因用于选择转化的细胞或组织。所述选择性标记基因包括但不限于,编码抗生素抗性的基因(如编码新霉素磷酸转移酶II(NPT))和潮霉素磷酸转移酶(HPT)的基因,编码磷酸甘露糖异构酶(PMI)的基因,以及赋予除草剂抗性的基因如草胺磷、溴苯腈、咪唑啉酮类和2,4-二氯苯氧乙酸酯(2,4-D)。本发明中,“试剂盒”可包括本发明描述的基因组修饰系统与以下的任一种或全部:测定试剂、缓冲液、探针和/或引物、和无菌盐水或另一种药学上可接受的乳剂和悬液基底。此外,试剂盒可包括含有用于实践本发明描述的方法的用法说明(例如,操作方案)的说明性材料。转化方案以及将核苷酸序列导入植物的方案依定向转化的植物或植物细胞类型而异,即单子叶植物或双子叶植物。将核苷酸序列导入植物细胞并随后插入植物基因组中的适合方法包括但不限于,农杆菌介导的转化、微量发射轰击、直接将DNA摄入原生质体、电穿孔或晶须硅介导的DNA导入。已经转化的细胞可按照常规的方式生长成植物。这些植物被培育,用相同的转化株或不同的转化株授粉,得到的杂交体表达所需的被鉴定的表现型特征。可培育二代或多代以保证稳定地保持和遗传所需表现型特征的表达,然后收获可保证得到所需表现型特征表达的种子。本发明提供了一种基因组编辑系统及其用途,具有以下优点:1、转录起始位点无限制:使用II型启动子驱动sgRNA表达,消除了对于转录起始位点的碱基限制。2、指导切割特异组织:大多数特异表达的启动子属于II型启动子,若使用该类型启动子驱动sgRNA的表达,有利于实现特异组织的基因编辑。3、拓宽sgRNA可利用的启动子范围:使用II型启动子转录sgRNA,拓宽了sgRNA可利用的启动子范围,同时消除了对于靶点第一位碱基的限制,扩大了靶点选择范围。4、简化载体:通过将sgRNA和Cas9整合在一个表达盒中,不仅可以减少载体构建过程中表达盒的数量、简化载体,还能避免在某些品种农杆菌中由于多个表达盒使用相同的启动子而发生重组导致部分元件的丢失的问题。5、编辑效率较高:本发明提供的由II型启动子驱动的基因编辑系统获得的编辑效率与常规体系相当,并且可以直接应用于实际生产中。附图说明图1为本发明基因组编辑系统及其用途的重组克隆载体DBN02-T构建流程图;图2为本发明基因组编辑系统及其用途的重组表达载体DBN100677构建流程图;图3为本发明基因组编辑系统及其用途的sgRNA与内含子不同嵌合方式的玉米悬浮细胞团的GUS染色效果图;图4为本发明基因组编辑系统及其用途的重组克隆剪刀载体DBN04-T构建流程图;图5为本发明基因组编辑系统及其用途的重组表达载体DBN100777构建流程图;图6为本发明基因组编辑系统及其用途的重组表达载体DBN100775构建流程图;图7为本发明基因组编辑系统及其用途的II型启动子驱动Cas9/sgRNA的玉米悬浮细胞团的GUS染色效果图;图8为本发明基因组编辑系统及其用途的重组表达载体DBN100000载体结构图;图9为本发明基因组编辑系统及其用途的靶点1载体DBN-A载体结构图;图10为本发明基因组编辑系统及其用途的靶点2载体DBN-B载体结构图;图11为本发明基因组编辑系统及其用途的重组表达载体DBN100778构建流程图;图12为本发明基因组编辑系统及其用途的靶点3载体DBN-C载体结构图;图13为本发明基因组编辑系统及其用途的靶点4载体DBN-D载体结构图。具体实施方式下面通过具体实施例进一步说明本发明基因组编辑系统及其用途的技术方案。第一实施例、sgRNA与内含子嵌合方式的筛选一、sgRNA与内含子在不同嵌合方式下组成的嵌合序列的获得和合成1、获得sgRNA与内含子在不同连接方式下组成的嵌合序列GUS蛋白的氨基酸序列如序列表中SEQIDNO:1所示,编码相应于所述GUS蛋白的氨基酸序列的GUS-01核苷酸序列如序列表中SEQIDNO:2所示,其中内含子iGUS-01序列如序列表中SEQIDNO:3所示;将sgRNA序列(如序列表中SEQIDNO:4所示)插入到所述内含子iGUS-01内部形成的嵌合序列iGUS-02(含酶切位点BsaI),如序列表中SEQIDNO:5所示,包含所述嵌合序列iGUS-02的GUS-02核苷酸序列如序列表中SEQIDNO:6所示;将所述内含子iGUS-01序列部分替换为sgRNA(如序列表中SEQIDNO:4所示)后形成的嵌合序列iGUS-03(含酶切位点BsaI),如序列表中SEQIDNO:7所示,包含所述嵌合序列iGUS-03的GUS-03核苷酸序列如序列表中SEQIDNO:8所示。2、合成上述核苷酸序列所述GUS-01核苷酸序列(如序列表中SEQIDNO:2所示)、所述GUS-02核苷酸序列(如序列表中SEQIDNO:6所示)和所述GUS-03核苷酸序列(如序列表中SEQIDNO:8所示)由南京金斯瑞生物科技有限公司合成;合成的所述GUS-01核苷酸序列的5’端还连接有KpnI酶切位点,所述GUS-01核苷酸序列的3’端还连接有SpeI酶切位点;合成的所述GUS-02核苷酸序列的5’端还连接有KpnI酶切位点,所述GUS-02核苷酸序列的3’端还连接有SpeI酶切位点;合成的所述GUS-03核苷酸序列的5’端还连接有KpnI酶切位点,所述GUS-03核苷酸序列的3’端还连接有SpeI酶切位点。二、重组表达载体的构建及重组表达载体转化农杆菌1、构建重组克隆载体将合成的GUS-02核苷酸序列连入克隆载体pGEM-T(Promega,Madison,USA,CAT:A3600)上,操作步骤按Promega公司产品pGEM-T载体说明书进行,得到重组克隆载体DBN02-T,其构建流程如图1所示(其中,Amp表示氨苄青霉素抗性基因;f1表示噬菌体f1的复制起点;LacZ为LacZ起始密码子;SP6为SP6RNA聚合酶启动子;T7为T7RNA聚合酶启动子;GUS-02为GUS-02核苷酸序列(SEQIDNO:6);MCS为多克隆位点)。然后将重组克隆载体DBN02-T用热激方法转化大肠杆菌T1感受态细胞(Transgen,Beijing,China,CAT:CD501),其热激条件为:50μl大肠杆菌T1感受态细胞、10μl质粒DNA(重组克隆载体DBN02-T),42℃水浴30秒;37℃振荡培养1小时(100rpm转速下摇床摇动),在表面涂有IPTG(异丙基硫代-β-D-半乳糖苷)和X-gal(5-溴-4-氯-3-吲哚-β-D-半乳糖苷)的氨苄青霉素(100mg/L)的LB平板(胰蛋白胨10g/L、酵母提取物5g/L、NaCl10g/L、琼脂15g/L,用NaOH调pH至7.5)上生长过夜。挑取白色菌落,在LB液体培养基(胰蛋白胨10g/L、酵母提取物5g/L、NaCl10g/L、氨苄青霉素100mg/L,用NaOH调pH至7.5)中于温度37℃条件下培养过夜。碱法提取其质粒:将菌液在12000rpm转速下离心1min,去上清液,沉淀菌体用100μl冰预冷的溶液I(25mMTris-HCl、10mMEDTA(乙二胺四乙酸)、50mM葡萄糖,pH8.0)悬浮;加入200μl新配制的溶液II(0.2MNaOH、1%SDS(十二烷基硫酸钠)),将管子颠倒4次,混合,置冰上3-5min;加入150μl冰冷的溶液III(3M醋酸钾、5M醋酸),立即充分混匀,冰上放置5-10min;于温度4℃、转速12000rpm条件下离心5min,在上清液中加入2倍体积无水乙醇,混匀后室温放置5min;于温度4℃、转速12000rpm条件下离心5min,弃上清液,沉淀用浓度(V/V)为70%的乙醇洗涤后晾干;加入30μl含RNase(20μg/ml)的TE(10mMTris-HCl、1mMEDTA,pH8.0)溶解沉淀;于温度37℃下水浴30min,消化RNA;于温度-20℃保存备用。提取的质粒经KpnI和SpeI酶切鉴定后,对阳性克隆进行测序验证,结果表明重组克隆载体DBN02-T中插入的所述GUS-02核苷酸序列为序列表中SEQIDNO:6所示的核苷酸序列,即GUS-02核苷酸序列正确插入。按照上述构建重组克隆载体DBN02-T的方法,将合成的所述GUS-01核苷酸序列连入克隆载体pGEM-T上,得到重组克隆载体DBN01-T,其中,GUS-01为GUS-01核苷酸序列(SEQIDNO:3)。酶切和测序验证重组克隆载体DBN01-T中所述GUS-01核苷酸序列正确插入。按照上述构建重组克隆载体DBN02-T的方法,将合成的所述GUS-03核苷酸序列连入克隆载体pGEM-T上,得到重组克隆载体DBN03-T,其中,GUS-03为GUS-03核苷酸序列(SEQIDNO:8)。酶切和测序验证重组克隆载体DBN03-T中所述GUS-03核苷酸序列正确插入。2、构建重组表达载体将pCAMBIA2300(CAMBIA机构可以提供)载体进行改造,利用常规的酶切方法构建载体是本领域技术人员所熟知的,通过点突变去掉pCAMBIA2300载体上的BsaI位点,同时将卡那霉素表达盒去掉,得到pDBN骨架载体。向所述pDBN骨架载体引入花椰菜花叶病毒35S启动子和终止子以及PMI表达盒(prUbi+PMI+tNos),得到表达载体DBN-PMI,用于下述载体构建。用限制性内切酶KpnI和SpeI分别酶切重组克隆载体DBN02-T和表达载体DBN-PMI,将切下的GUS-02核苷酸序列片段插到表达载体DBN-PMI的KpnI和SpeI位点之间,利用常规的酶切方法构建载体是本领域技术人员所熟知的,构建成重组表达载体DBN100677,其构建流程如图2所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);GUS-02:GUS-02核苷酸序列(SEQIDNO:6);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);LB:左边界)。将重组表达载体DBN100677用热激方法转化大肠杆菌T1感受态细胞,其热激条件为:50μl大肠杆菌T1感受态细胞、10μl质粒DNA(重组表达载体DBN100677),42℃水浴30秒;37℃振荡培养1小时(100rpm转速下摇床摇动);然后在含50mg/L卡那霉素(Kanamycin)的LB固体平板(胰蛋白胨10g/L、酵母提取物5g/L、NaCl10g/L、琼脂15g/L,用NaOH调pH至7.5)上于温度37℃条件下培养12小时,挑取白色菌落,在LB液体培养基(胰蛋白胨10g/L、酵母提取物5g/L、NaCl10g/L、卡那霉素50mg/L,用NaOH调pH至7.5)中于温度37℃条件下培养过夜。碱法提取其质粒。将提取的质粒用限制性内切酶KpnI和SpeI酶切后鉴定,并将阳性克隆进行测序鉴定,结果表明重组表达载体DBN100677在KpnI和SpeI位点间的核苷酸序列为序列表中SEQIDNO:6所示核苷酸序列,即GUS-02核苷酸序列。按照上述构建重组表达载体DBN100677的方法,将KpnI和SpeI酶切重组克隆载体DBN01-T切下的所述GUS-01核苷酸序列插入表达载体DBN-PMI,得到重组表达载体DBN100774。酶切和测序验证重组表达载体DBN100774中的核苷酸序列含有为序列表中SEQIDNO:3所示核苷酸序列,即GUS-01核苷酸序列,所述GUS-01核苷酸序列可以连接所述pr35S启动子和t35S终止子。按照上述构建重组表达载体DBN100677的方法,将KpnI和SpeI酶切重组克隆载体DBN03-T切下的所述GUS-03核苷酸序列插入表达载体DBN-PMI,得到重组表达载体DBN100678。酶切和测序验证重组表达载体DBN100678中的核苷酸序列含有为序列表中SEQIDNO:8所示核苷酸序列,即GUS-03核苷酸序列,所述GUS-03核苷酸序列可以连接所述pr35S启动子和t35S终止子。3、重组表达载体转化农杆菌对己经构建正确的重组载体DBN100677、DBN100678和DBN100774用液氮法转化到农杆菌LBA4404(Invitrgen,Chicago,USA,CAT:18313-015)中,其转化条件为:100μl农杆菌LBA4404、3μl质粒DNA(重组表达载体);置于液氮中10min,37℃温水浴10min;将转化后的农杆菌LBA4404接种于LB试管中于温度28℃、转速为200rpm条件下培养2小时,涂于含50mg/L的利福平(Rifampicin)和100mg/L的卡那霉素的LB平板上直至长出阳性单克隆,挑取单克隆培养并提取其质粒,用限制性内切酶对重组表达载体DBN100677、DBN100678和DBN100774酶切后进行酶切验证,结果表明重组表达载体DBN100677、DBN100678和DBN100774结构完全正确。三、转化的玉米悬浮细胞系的获得1、玉米幼胚的剥取将玉米品种齐319(Q319)的种子播种于温室或大田中,待玉米植株长成授粉后7-12天,收获玉米雌穗,用浓度为75%(v/v)的酒精浸泡5min后剥取幼胚,挑选长度为0.6-1.2mm的透明幼胚备用。2、初生愈伤组织的获得将上述剥取的玉米幼胚接种到愈伤诱导培养基(MS盐4.3g/L、N6维生素、脯氨酸0.5g/L、水解酪蛋白0.1g/L、天冬氨酸0.2g/L、蔗糖30g/L、麦草畏(Dicamba)2mg/L、2,4-D1.5mg/L、毒莠定(Picloram)2.2mg/L、激动素(KT)0.3mg/L、苄氨基嘌呤(BAP)0.02mg/L、植物凝胶3g/L,pH5.8)上进行愈伤组织的诱导,于温度28℃的条件下暗培养10-14天,优选为14天,获得表面突起明显、颜色鲜嫩、生长速度提高的初生愈伤组织。3、制备玉米悬浮细胞系玉米悬浮细胞系的起始:将获得的所述初生愈伤组织转移至装有75ml悬浮培养液SUS-1(MS盐4.3g/L、MS维生素、脯氨酸3g/L、水解酪蛋白1.5g/L、蔗糖45g/L、Dicamba1.5mg/L、2,4-D1mg/L、Picloram5mg/L,pH5.8)的无菌三角瓶(150ml)中,每个三角瓶中接种5-8粒所述初生愈伤组织,在温度为28-30℃暗培养条件下,将密封的上述三角瓶置于转速为100-150r/min恒温摇床上震荡培养。玉米悬浮细胞系的获得:所述初生愈伤组织悬浮培养10-25天后进行第一次继代,优选为20天,继代时剔除褐化死亡的愈伤组织,在原三角瓶中保留生长旺盛、质地坚硬、色泽鲜黄的新生愈伤组织,且在保留一半原有悬浮培养液SUS-1的基础上,加入新的玉米悬浮培养液SUS-1至三角瓶75ml刻度处,在温度为28-30℃暗培养条件下,将密封的上述三角瓶置于转速为100-150r/min恒温摇床上震荡培养,以便获得大量的愈伤组织;之后每周继代一次,每次继代均需剔除褐化死亡的愈伤组织,保留新生愈伤组织,且每次继代均保留一半原有悬浮培养液SUS-1,并加入新的悬浮培养液SUS-1至三角瓶75ml刻度处,在温度为28-30℃暗培养条件下,将所述三角瓶置于转速为100-150r/min恒温摇床上震荡培养。所述初生愈伤组织悬浮培养35-45天后,优选为40天,逐渐产生结构松散的多个悬浮细胞团,将所述悬浮细胞团转移至装有75ml悬浮培养液SUS-2(MS盐4.3g/L、MS维生素、脯氨酸3g/L、水解酪蛋白1.5g/L、蔗糖30g/L、Dicamba1.5mg/L,pH5.8)新的无菌三角瓶(150ml)中,每个三角瓶中接种20-30粒所述悬浮细胞团,在温度为28-30℃暗培养条件下,将密封的上述三角瓶置于转速为100-150r/min恒温摇床上震荡培养5-7天后,多个所述悬浮细胞团即可形成分散良好、增值迅速的玉米悬浮细胞系。4、农杆菌介导的玉米悬浮细胞系遗传转化预培养:将所述玉米悬浮细胞系中制备的所述悬浮细胞团接种到PRE培养基(MS盐4.3g/L、N6维生素、脯氨酸0.5g/L、水解酪蛋白0.1g/L、天冬氨酸0.79g/L、蔗糖30g/L、2,4-D2mg/L、植物凝胶3g/L,pH5.8)上,在温度为28-30℃暗培养条件下预培养4-7天备用,优选为5天。预处理:将预培养后的所述玉米悬浮细胞团进行农杆菌侵染前预处理,即60℃烘箱热处理5分钟后超声波处理5分钟,预处理后的所述玉米悬浮细胞团用于后续农杆菌侵染。农杆菌菌液的制备:分别挑取含有DBN100677、DBN100678和DBN100774的农杆菌单菌落,用枪头分别在含有50mg/L卡那霉素的固体YP培养板(胰蛋白胨10g/L、酵母提取物5g/L、NaCl5g/L、卡那霉素50mg/L、琼脂粉15g/L)上划线,在温度28℃黑暗条件下培养2-3天。刮取所述固体YP培养板上的菌斑,在新的固体YP培养板上再培养1天,刮取共计培养3-4天的菌落,悬浮在5mL液体INF侵染培养基(MS盐4.3g/L、N6维生素、Dicamba1.5mg/L、脯氨酸0.5mg/L、蔗糖30g/L、肌醇0.1g/L、乙酰丁香酮(AS)50mg/L,pH5.4)中,将农杆菌菌液混匀,将其浓度调整为OD660=0.5-0.6。农杆菌侵染玉米悬浮细胞团:将上述预处理后的所述玉米悬浮细胞团分别浸泡于所述农杆菌菌液(20-30ml)中5-25min,优选25min;侵染结束后将农杆菌菌液吸出,然后将所述玉米悬浮细胞团分别转移至滤纸上,并在超净工作台中风干5-20min。农杆菌与玉米悬浮细胞团共培养:将侵染风干后的所述玉米悬浮细胞团分别转移到共培养培养基(MS盐4.3g/L、N6维生素、脯氨酸0.5g/L、蔗糖30g/L、2,4-D2mg/L、乙酰丁香酮50mg/L、山梨醇36g/L、植物凝胶3g/L,pH5.4)上,在温度20-28℃条件下暗培养至少2天,优选为5天。由上述步骤获得转入DBN100677的玉米悬浮细胞系、转入DBN100678的玉米悬浮细胞系和转入DBN100774的玉米悬浮细胞系。四、sgRNA与内含子嵌合方式的功能验证分别取转入DBN100678的玉米悬浮细胞团和转入DBN100677的玉米悬浮细胞团作为样品,参照Jefferson等(JeffersonR.A.,BurgessS.M.,HirshD.Beta-glucuronidasefromEscherichiacoliasagenefusionmarker.Proc.Natl.Acad.Sci.,1986,83:8447-8454)的方法,通过在染色溶液中37℃密封染色两天,从组织化学上检验GUS的表达方式。所述染色溶液的主要成分为:0.1MNaPO4、0.01MEDTA(pH8.0)、0.5mM铁氰化钾、0.5mM亚铁氰化钾、0.5mg/ml5-溴-4-氯-3-吲哚-β-D-葡糖苷酸(X-gluc),其中X-gluc可以被转基因细胞中产生的GUS蛋白原位分解并生成蓝色沉淀,从而可以定位GUS蛋白的表达。染色后的玉米悬浮细胞团在解剖显微镜下检查和照相。同时以转入DBN100774的玉米悬浮细胞团作为对照,按照上述方法进行检测分析,以上实验设3次重复。玉米悬浮细胞团的GUS染色的实验结果如图3所示。实验结果表明作为对照的转入DBN100774的玉米悬浮细胞团染色面积最大,表明GUS蛋白表达完全;转入GUS-02的玉米悬浮细胞团染色面积较小,表明以sgRNA插入到内含子内部的嵌合方式,有部分GUS蛋白可以表达,仅降低了GUS蛋白表达的强度,但并不影响部分内含子剪切;转入GUS-03的玉米悬浮细胞团基本上不显色,表明将内含子序列部分替换为sgRNA的嵌合方式,影响了内含子的剪切从而使GUS蛋白完全不表达。上述筛选结果表明:sgRNA插入到内含子内部的嵌合方式(GUS-02核苷酸序列)不影响部分内含子剪切,sgRNA能够被转录,且可以由II型启动子驱动表达。第二实施例、II型启动子驱动CRISPR/Cas9系统的活性检测一、II型启动子驱动CRISPR/Cas9系统的载体构建1、构建重组克隆剪刀载体iGUS-02-Cas9核苷酸序列(如序列表中SEQIDNO:14所示,其中Cas9核苷酸序列如序列表中SEQIDNO:15所示)由南京金斯瑞生物科技有限公司合成。将合成的iGUS-02-Cas9核苷酸序列连入克隆载体pGEM-T(Promega,Madison,USA,CAT:A3600)上,操作步骤按Promega公司产品pGEM-T载体说明书进行,得到重组克隆剪刀载体DBN04-T,其构建流程如图4所示(其中,Amp表示氨苄青霉素抗性基因;f1表示噬菌体f1的复制起点;LacZ为LacZ起始密码子;SP6为SP6RNA聚合酶启动子;T7为T7RNA聚合酶启动子;iGUS-02-Cas9为嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:14;Cas9氨基酸序列如SEQIDNO:16所示);MCS为多克隆位点)。按照第一实施例二中1的方法,将重组克隆剪刀载体DBN04-T用热激方法转化大肠杆菌T1感受态细胞,并对阳性克隆进行测序验证,结果表明重组克隆剪刀载体DBN04-T中插入的所述iGUS-02-Cas9核苷酸序列为序列表中SEQIDNO:14所示的核苷酸序列,即iGUS-02-Cas9核苷酸序列正确插入。2、构建无靶点重组表达载体向按照第一实施例二中2所述表达载体DBN-PMI引入GUUS表达盒(pr35S-02+GUUS+tNos),得到表达载体DBN-PMI-GUUS,用于下述载体构建。用限制性内切酶KpnI和SpeI分别酶切重组克隆剪刀载体DBN04-T和表达载体DBN-PMI-GUUS,将切下的iGUS-02-Cas9核苷酸序列片段插到表达载体DBN-PMI-GUUS中,利用常规的酶切方法构建载体是本领域技术人员所熟知的,构建成重组表达载体DBN100777,其构建流程如图5所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);iGUS-02-Cas9:嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:14);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);pr35S-02:花椰菜花叶病毒35S启动子(SEQIDNO:17);GUUS:含有45CS1靶点序列的GUS基因(如SEQIDNO:18所示);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);LB:左边界)。按照第一实施例二中2的方法,将重组表达载体DBN100777用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,将提取的质粒用限制性内切酶KpnI和SpeI酶切后鉴定,对阳性克隆进行测序验证,结果表明重组表达载体DBN100777中插入的所述iGUS-02-Cas9核苷酸序列为序列表中SEQIDNO:14所示的核苷酸序列,即iGUS-02-Cas9核苷酸序列正确插入。3、靶点的引物设计与靶点载体构建根据Cas9蛋白识别PAM序列(NGG/NGGNG)的原则,在CRISPRDirect网站(http://crispr.dbcls.jp/)筛选到来源于玉米的一个靶点序列(20bp),即45CS1靶点序列,如SEQIDNO:19所示。45CS1正向引物:ggtctccaaacCCCAAGAGCGGGCGAGGTAG,如SEQIDNO:20所示;45CS1反向引物:ggtctcgaaaacCTACCTCGCCCGCTCTTGGG,如SEQIDNO:21所示;其中,上述引物5’端的粗体小写字母为保护碱基,斜体小写字母为酶切位点BsaI,带下划线的小写字母为酶切位点BsaI的粘性末端,正常字体大写字母为45CS1靶点序列或其反向互补序列。所述45CS1正向引物和所述45CS1反向引物分别包括45CS1靶点序列,二者反向互补,互为模板,用Pfu酶(NEB)进行PCR扩增,PCR体系如下:PCR反应条件为:98℃预变性30s,然后进入下列循环:98℃变性10s,56-60℃退火30s,72℃延伸30s/kb,共30-32个循环,最后72℃延伸5-10min;于4℃保存。使用过柱纯化试剂盒(购自北京全式金生物技术有限公司)对上述PCR产物过柱纯化,具体方法参考其产品说明书;BsaI酶切PCR产物和表达载体DBN100777,切胶回收对应酶切产物后,将酶切后的表达载体DBN100777产物和PCR产物按照1:10的比例用T4连接酶于温度16℃下连接30min,利用常规的酶切方法构建载体是本领域技术人员所熟知的,构建成靶点载体DBN100775,其构建流程图如图6所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);45CS1:45CS1靶位点序列(SEQIDNO:19);45CS1+iGUS-02-Cas9:包含45CS1靶点序列的嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:22);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);pr35S-02:花椰菜花叶病毒35S启动子(SEQIDNO:17);GUUS:含有45CS1靶点序列的GUS基因(如SEQIDNO:18所示);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);LB:左边界)。按照第一实施例二中2的方法,将重组表达载体DBN100775用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,提取的质粒经KpnI和AscI酶切鉴定后,对阳性克隆进行测序验证,结果表明靶点载体DBN100775中GUS靶位点序列正确插入。4、重组表达载体转化农杆菌对己经构建正确的重组表达载体DBN100777和DBN100775用液氮法转化到农杆菌LBA4404(Invitrgen,Chicago,USA,CAT:18313-015)中,其转化条件为:100μl农杆菌LBA4404、3μl质粒DNA(重组表达载体);置于液氮中10min,37℃温水浴10min;将转化后的农杆菌LBA4404接种于LB试管中于温度28℃、转速为200rpm条件下培养2小时,涂于含50mg/L的利福平和100mg/L的卡那霉素的LB平板上直至长出阳性单克隆,挑取单克隆培养并提取其质粒,用限制性内切酶对重组表达载体DBN100777和DBN100775酶切后进行酶切验证,结果表明重组表达载体DBN100777和DBN100775结构完全正确。二、瞬时转化的玉米悬浮细胞系的获得玉米悬浮细胞系的获得及农杆菌介导的玉米悬浮细胞系的遗传转化如第一实施例三中所述。由此获得转入DBN100777的玉米悬浮细胞系、转入DBN100775的玉米悬浮细胞系和转入DBN100774的玉米悬浮细胞系。三、II型启动子驱动CRISPR/Cas9系统的活性检测按照第一实施例四中所述的方法,检测转入DBN100775的玉米悬浮细胞系的GUS活性,同时以转入DBN100777的玉米悬浮细胞团作为阴性对照,以转入DBN100774的玉米悬浮细胞团作为阳性对照,按照上述方法进行检测分析,以上实验设3次重复。玉米悬浮细胞团的GUS染色的实验结果如图7所示:转入DBN100775的玉米悬浮细胞团染色面积较大,基本上与阳性对照染色程度相当,而阴性对照则完全不显色。上述实验结果表明:转入DBN100775的玉米悬浮细胞团中Cas9蛋白能够被正常翻译且sgRNA能够被转录,两者相互作用,剪切GUUS之间的45CS1靶点,使得GUUS恢复为GUS蛋白,并可以被染色。由此可见,当靶点和sgRNA采用插入到内含子内部的嵌合方式,并有效连接于Cas9蛋白时,在II型启动子的驱动下,Cas9蛋白能够被正常翻译且sgRNA能够被转录,两者相互作用,剪切靶点位置,即II型启动子驱动下的Cas9/sgRNA(靶点和sgRNA插入到内含子内部的嵌合方式)系统能够正常行使剪切和/或编辑功能。第三实施例、稳定转化后验证组成型启动子驱动CRISPR/Cas9系统的活性一、水稻基因组靶点的选择根据Cas9蛋白识别PAM序列(NGG/NGGNG)的原则,在CRISPR-Plant网站(http://www.genome.arizona.edu/crispr/CRISPRsearch.html)筛选到来源于水稻的2个靶点序列,具体信息如下:靶点1序列:TTTACTGGGAGTTATGAAAC,如SEQIDNO:23所示;靶点2序列:GGACGAGTCCAGATGCACCG,如SEQIDNO:24所示。二、组成型启动子驱动CRISPR/Cas9系统的载体构建1、构建无靶点重组表达载体向按照第一实施例二中2所述pDBN骨架载体引入花椰菜花叶病毒35S启动子和终止子以及Hpt表达盒(prUbi+Hpt+tNos),得到表达载体DBN-Hpt,用于下述载体构建。用限制性内切酶KpnI和SpeI分别酶切第二实施例一中1所述重组克隆剪刀载体DBN04-T和表达载体DBN-Hpt,将切下的iGUS-02-Cas9核苷酸序列片段插到表达载体DBN-Hpt中,利用常规的酶切方法构建载体是本领域技术人员所熟知的,构建成重组表达载体DBN100000,其载体结构如图8所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);iGUS-02-Cas9:嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:14);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);Hpt:潮霉素磷酸转移酶基因(SEQIDNO:25);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);LB:左边界)。按照第一实施例二中2的方法,将重组表达载体DBN100000用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,将提取的质粒用限制性内切酶KpnI和AscI酶切后鉴定,对阳性克隆进行测序验证,结果表明重组表达载体DBN100000中插入的所述iGUS-02-Cas9核苷酸序列为序列表中SEQIDNO:14所示的核苷酸序列,即iGUS-02-Cas9核苷酸序列正确插入。2、靶点的引物设计与靶点载体构建选择靶点1序列,如SEQIDNO:23所示;因此靶点1正向引物:ggtctccaaacTTTACTGGGAGTTATGAAAC,如SEQIDNO:26所示;靶点1反向引物:ggtctcgaaaacGTTTCATAACTCCCAGTAAA,如SEQIDNO:27所示;选择靶点2序列,如SEQIDNO:24所示;因此靶点2正向引物:ggtctccaaacGGACGAGTCCAGATGCACCG,如SEQIDNO:28所示;靶点2反向引物:ggtctcgaaaacCGGTGCATCTGGACTCGTCC,如SEQIDNO:29所示;其中,上述引物5’端的粗体小写字母为保护碱基,斜体小写字母为酶切位点BsaI,带下划线的小写字母为酶切位点BsaI的粘性末端,正常字体大写字母为靶点序列或其反向互补序列。按照第二实施例一中3的方法,构建成靶点1载体DBN-A,其载体结构如图9所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);靶点1:靶点1序列(SEQIDNO:23);靶点1+iGUS-02-Cas9:包含靶点1序列的嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:30);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);Hpt:潮霉素磷酸转移酶基因(SEQIDNO:25);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);LB:左边界)。按照第二实施例一中3的方法,构建成靶点2载体DBN-B,其载体结构如图10所示(Kan:卡那霉素基因;RB:右边界;pr35S-01:花椰菜花叶病毒35S启动子(SEQIDNO:9);靶点2:靶点2序列(SEQIDNO:24);靶点2+iGUS-02-Cas9:包含靶点2序列的嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:31);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);Hpt:潮霉素磷酸转移酶基因(SEQIDNO:25);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);LB:左边界)。按照第一实施例二中2的方法,将靶点1载体DBN-A和靶点2载体DBN-B分别用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,提取的质粒经KpnI和AscI酶切鉴定后,分别对阳性克隆进行测序验证,结果表明靶点1载体DBN-A和靶点2载体DBN-B中靶点1序列和靶点2序列均正确插入。3、重组表达载体转化农杆菌对己经构建正确的靶点1载体DBN-A和靶点2载体DBN-B用液氮法转化到农杆菌LBA4404(Invitrgen,Chicago,USA,CAT:18313-015)中,其转化条件为:100μl农杆菌LBA4404、3μl质粒DNA(重组表达载体);置于液氮中10min,37℃温水浴10min;将转化后的农杆菌LBA4404接种于LB试管中于温度28℃、转速为200rpm条件下培养2小时,涂于含50mg/L的利福平和100mg/L的卡那霉素的LB平板上直至长出阳性单克隆,挑取单克隆培养并提取其质粒,用限制性内切酶对靶点1载体DBN-A和靶点2载体DBN-B酶切后进行酶切验证,结果表明靶点1载体DBN-A和靶点2载体DBN-B结构完全正确。三、获得转化的水稻植株对于农杆菌介导的水稻转化,简要地,把水稻品种华占种子(华占水稻材料由北京金色农华种业有限公司提供)接种在诱导培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、麦芽糖30g/L、2,4-二氯苯氧乙酸(2,4-D)2mg/L、植物凝胶3g/L,pH5.8)上,从水稻成熟胚诱导出愈伤组织(步骤1:愈伤诱导步骤),之后,优选愈伤组织,用农杆菌悬浮液接触愈伤组织,其中农杆菌能够将所述靶点1载体DBN-A和所述靶点2载体DBN-B分别传递至愈伤组织上的至少一个细胞(步骤2:侵染步骤)。在此步骤中,愈伤组织优选地浸入农杆菌悬浮液(OD660=0.3,侵染培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、蔗糖30g/L、葡萄糖10g/L、乙酰丁香酮(AS)40mg/L、2,4-二氯苯氧乙酸(2,4-D)2mg/L、pH5.4))中以启动接种。愈伤组织与农杆菌共培养一段时期(3天)(步骤3:共培养步骤)。优选地,愈伤组织在侵染步骤后在固体培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、蔗糖30g/L、葡萄糖10g/L、乙酰丁香酮(AS)40mg/L、2,4-二氯苯氧乙酸(2,4-D)2mg/L、植物凝胶3g/L,pH5.8)上培养。在此共培养阶段后,有一个“恢复”步骤。在“恢复”步骤中,恢复培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、蔗糖30g/L、2,4-二氯苯氧乙酸(2,4-D)2mg/L、植物凝胶3g/L,pH5.8)中至少存在一种己知抑制农杆菌生长的抗生素(头孢霉素150-250mg/L),不添加植物转化体的选择剂(步骤4:恢复步骤)。优选地,愈伤组织在有抗生素但没有选择剂的固体培养基上培养,以消除农杆菌并为侵染细胞提供恢复期。接着,接种的愈伤组织在含选择剂(潮霉素)的培养基上培养并选择生长着的转化愈伤组织(步骤5:选择步骤)。优选地,愈伤组织在有选择剂的筛选固体培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、蔗糖5g/L、潮霉素50mg/L、2,4-二氯苯氧乙酸(2,4-D)2mg/L、植物凝胶3g/L,pH5.8)上培养,导致转化的细胞选择性生长。然后,愈伤组织再生成植物(步骤6:再生步骤),优选地,在含选择剂的培养基上生长的愈伤组织在固体培养基(N6分化培养基和MS生根培养基)上培养以再生植物。筛选得到的抗性愈伤组织转移到所述N6分化培养基(N6盐3.1g/L、N6维他命、干酪素300mg/L、蔗糖20g/L、潮霉素50mg/L、6-苄氨基腺嘌呤2mg/L、奈乙酸1mg/L、植物凝胶3g/L,pH5.8)上,25℃下培养分化。分化出来的小苗转移到所述MS生根培养基(MS盐2.15g/L、MS维他命、干酪素300mg/L、蔗糖15g/L、植物凝胶3g/L,pH5.8)上,25℃下培养至约10cm高,移至温室培养。在温室中,每天于28℃下培养,获得转化的T0水稻植株。四、稳定转化后组成型启动子驱动CRISPR/Cas9系统的功能验证测序结果如表1所示,获得靶点1突变体DBN-A水稻T0植株数为25株,其突变效率(突变效率=对应突变体数量/突变体T0植株总数×100%)为35.2%;获得靶点2突变体DBN-B水稻T0植株数为21株,其突变效率为29.5%。表1、组成型启动子驱动CRISPR/Cas9系统在稳定转化后的水稻基因组上应用的实验结果载体/项目对应突变体数(株)突变效率(%)DBN-A(靶点1)2516.2DBN-B(靶点2)2170常规CRISPR/Cas9载体(靶点1)3219常规CRISPR/Cas9载体(靶点2)2581上述实验结果表明:稳定转化所述靶点1载体DBN-A和所述靶点2载体DBN-B的水稻植株中,sgRNA能够通过组成型启动子转录,并可与Cas9蛋白结合行使编辑功能以获得突变体,且突变效率与常规CRISPR/Cas9载体系统相当。由此可见,在稳定转化后的转化体中,当靶点和sgRNA采用插入到内含子内部的嵌合方式,并有效连接于Cas9蛋白时,在II型组成型启动子的驱动下,Cas9蛋白能够被正常翻译且sgRNA能够被转录,两者相互作用,剪切靶点位置,即II型组成型启动子驱动下的Cas9/sgRNA(靶点和sgRNA插入到内含子内部的嵌合方式)系统能够正常行使剪切和/或编辑功能,且突变效率与常规CRISPR/Cas9载体系统相当。第四实施例、稳定转化后验证特异性启动子驱动CRISPR/Cas9系统的活性一、玉米基因组靶点的选择根据Cas9蛋白识别PAM序列(NGG/NGGNG)的原则,在CRISPR-Plant网站(http://www.genome.arizona.edu/crispr/CRISPRsearch.html)筛选到来源于玉米一个基因上的2个靶点序列,具体信息如下:靶点3:ATCATACCTGAGCAGCCTCC,如SEQIDNO:32所示;靶点4:CGAACGACCTCGATGTGCAC,如SEQIDNO:33所示。二、特异性启动子驱动CRISPR/Cas9系统的载体构建1、构建无靶点重组表达载体将第二实施例一中2所述DBN100777的花椰菜花叶病毒启动子pr35S-01更换为花粉特异表达启动子prAmy,构建成重组表达载体DBN100778,其载体结构如图11所示(Kan:卡那霉素基因;RB:右边界;prAmy:玉米α淀粉酶基因启动子(SEQIDNO:34);iGUS-02-Cas9:嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:14);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);pr35S-02:花椰菜花叶病毒35S启动子(SEQIDNO:17);GUUS:含有45CS1靶点序列的GUS基因(如SEQIDNO:18所示);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);LB:左边界)。按照第一实施例二中2的方法,将重组表达载体DBN100778用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,将提取的质粒用限制性内切酶BamHI和KpnI酶切后鉴定,对阳性克隆进行测序验证,结果表明重组表达载体DBN100778构建正确。2、靶点的引物设计与靶点载体构建选择靶点3序列,如SEQIDNO:32所示;因此靶点3正向引物:ggtctccaaacATCATACCTGAGCAGCCTCC,如SEQIDNO:35所示;靶点3反向引物:ggtctcgaaaacGGAGGCTGCTCAGGTATGAT,如SEQIDNO:36所示;选择靶点4序列,如SEQIDNO:33所示;因此靶点4正向引物:ggtctccaaacCGAACGACCTCGATGTGCAC,如SEQIDNO:37所示;靶点4反向引物:ggtctccaaaacGTGCACATCGAGGTCGTTCG,如SEQIDNO:38所示;其中,上述引物5’端的粗体小写字母为保护碱基,斜体小写字母为酶切位点BsaI,带下划线的小写字母为酶切位点BsaI的粘性末端,正常字体小写字母为靶点序列或其反向互补序列。按照第二实施例一中3的方法,构建成靶点3载体DBN-C,其载体结构如图12所示(Kan:卡那霉素基因;RB:右边界;prAmy:玉米α淀粉酶基因启动子(SEQIDNO:34);靶点3:靶点3序列(SEQIDNO:32);靶点3+iGUS-02-Cas9:包含靶点3序列的嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:39);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);pr35S-02:花椰菜花叶病毒35S启动子(SEQIDNO:17);GUUS:含有45CS1靶点序列的GUS基因(如SEQIDNO:18所示);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);LB:左边界)按照第二实施例一中3的方法,构建成靶点4载体DBN-D,其载体结构如图13所示(Kan:卡那霉素基因;RB:右边界;prAmy:玉米α淀粉酶基因启动子(SEQIDNO:34);靶点4:靶点4序列(SEQIDNO:33);靶点4+iGUS-02-Cas9:包含靶点4序列的嵌合序列iGUS-02+Cas9核苷酸序列(SEQIDNO:40);t35S:花椰菜花叶病毒35S终止子(SEQIDNO:10);pr35S-02:花椰菜花叶病毒35S启动子(SEQIDNO:17);GUUS:含有45CS1靶位点序列的GUS基因(如SEQIDNO:18所示);tNos:胭脂碱合成酶基因的终止子(SEQIDNO:13);prUbi:玉米泛素(Ubiquitin)基因启动子(SEQIDNO:11);PMI:磷酸甘露糖异构酶基因(SEQIDNO:12);LB:左边界)按照第一实施例二中2的方法,将靶点3载体DBN-C和靶点4载体DBN-D分别用热激方法转化大肠杆菌T1感受态细胞,碱法提取其质粒,提取的质粒经BamHI和KpnI酶切鉴定后,分别对阳性克隆进行测序验证,结果表明靶点3载体DBN-C和靶点4载体DBN-D中靶点3序列和靶点4序列均正确插入。3、重组表达载体转化农杆菌对己经构建正确的靶点3载体DBN-C和靶点4载体DBN-D用液氮法转化到农杆菌LBA4404(Invitrgen,Chicago,USA,CAT:18313-015)中,其转化条件为:100μl农杆菌LBA4404、3μl质粒DNA(重组表达载体);置于液氮中10min,37℃温水浴10min;将转化后的农杆菌LBA4404接种于LB试管中于温度28℃、转速为200rpm条件下培养2小时,涂于含50mg/L的利福平和100mg/L的卡那霉素的LB平板上直至长出阳性单克隆,挑取单克隆培养并提取其质粒,用限制性内切酶对靶点3载体DBN-C和靶点4载体DBN-D酶切后进行酶切验证,结果表明靶点3载体DBN-C和靶点4载体DBN-D结构完全正确。三、获得转化的玉米植株可以采用下述方法吗?按照常规采用的农杆菌侵染法,将无菌培养的玉米品种综31(Z31)的幼胚与第四实施例二中3所述的重组表达载体转化的农杆菌共培养,以将第四实施例二中1和2构建的重组表达载体DBN100778、DBN-C和DBN-D中的T-DNA转入到玉米染色体组中,获得了转入DBN100778的玉米植株、转入DBN-C的玉米植株和转入DBN-D的玉米植株。对于农杆菌介导的玉米转化,简要地,从玉米中分离未成熟的幼胚,用农杆菌悬浮液接触幼胚,其中农杆菌能够将能够将重组表达载体DBN100778、DBN-C和DBN-D中的T-DNA传递至幼胚之一的至少一个细胞(步骤1:侵染步骤),在此步骤中,幼胚优选地浸入农杆菌悬浮液(OD660=0.4-0.6,侵染培养基(MS盐4.3g/L、MS维他命、干酪素300mg/L、蔗糖68.5g/L、葡萄糖36g/L、乙酰丁香酮(AS)40mg/L、2,4-二氯苯氧乙酸(2,4-D)1mg/L,pH5.3))中以启动接种。幼胚与农杆菌共培养一段时期(3天)(步骤2:共培养步骤)。优选地,幼胚在侵染步骤后在固体培养基(MS盐4.3g/L、MS维他命、干酪素300mg/L、蔗糖20g/L、葡萄糖10g/L、乙酰丁香酮(AS)100mg/L、2,4-二氯苯氧乙酸(2,4-D)1mg/L、琼脂8g/L,pH5.8)上培养。在此共培养阶段后,可以有一个选择性的“恢复”步骤。在“恢复”步骤中,恢复培养基(MS盐4.3g/L、MS维他命、干酪素300mg/L、蔗糖30g/L、2,4-二氯苯氧乙酸(2,4-D)1mg/L、琼脂8g/L,pH5.8)中至少存在一种己知抑制农杆菌生长的抗生素(头孢霉素),不添加植物转化体的选择剂(步骤3:恢复步骤)。优选地,幼胚在有抗生素但没有选择剂的固体培养基上培养,以消除农杆菌并为侵染细胞提供恢复期。接着,接种的幼胚在含选择剂(甘露糖)的培养基上培养并选择生长着的转化愈伤组织(步骤4:选择步骤)。优选地,幼胚在有选择剂的筛选固体培养基(MS盐4.3g/L、MS维他命、干酪素300mg/L、蔗糖5g/L、甘露糖12.5g/L、2,4-二氯苯氧乙酸(2,4-D)1mg/L、琼脂8g/L,pH5.8)上培养,导致转化的细胞选择性生长。然后,愈伤组织再生成植物(步骤5:再生步骤),优选地,在含选择剂的培养基上生长的愈伤组织在固体培养基(MS分化培养基和MS生根培养基)上培养以再生植物。筛选得到的抗性愈伤组织转移到所述MS分化培养基(MS盐4.3g/L、MS维他命、干酪素300mg/L、蔗糖30g/L、6-苄基腺嘌呤2mg/L、甘露糖5g/L、琼脂8g/L,pH5.8)上,25℃下培养分化。分化出来的小苗转移到所述MS生根培养基(MS盐2.15g/L、MS维他命、干酪素300mg/L、蔗糖30g/L、吲哚-3-乙酸1mg/L、琼脂8g/L,pH5.8)上,25℃下培养至约10cm高,移至温室培养至结实。在温室中,每天于28℃下培养16小时,再于20℃下培养8小时。四、稳定转化后特异性启动子驱动CRISPR/Cas9系统的功能验证由上述方法分别获得19个株系转入DBN-C的T0玉米植株和21个株系转入DBN-D的T0玉米植株。在温室中培养T0植株,分别收获每个株系的T1种子。将19个株系转入DBN-C的T0玉米植株和21个株系转入DBN-D的T0玉米植株的T1种子(每个株系播种20粒种子)在温室中进行小盆种植,播种后10天,获得19个株系转入DBN-C的T1玉米植株和21个株系转入DBN-D的T1玉米植株,分别提取T1玉米植株的叶片DNA并通过测序进行突变效率的检测。测序结果如表2所示,3个株系转入DBN-C的T0玉米植株发生了突变,其突变效率(突变效率=对应突变的转入DBN-C的T0玉米植株的株系数/转入DBN-C的T0玉米植株的株系总数×100%)为21%;2个株系转入DBN-D的T0玉米植株发生了突变,其突变效率为9.5%。表2、特异性启动子驱动CRISPR/Cas9系统在稳定转化后的玉米基因组上应用的实验结果载体/项目对应突变体数(株系)总数(株系)突变效率(%)DBN-C(靶点3)41921DBN-D(靶点4)2219.5上述实验结果表明:稳定转化所述靶点3载体DBN-C和所述靶点4载体DBN-D的玉米植株中,sgRNA能够通过特异性启动子转录,并可与Cas9蛋白结合行使编辑功能以获得突变体。由此可见,在稳定转化后的转化体中,当靶点和sgRNA采用插入到内含子内部的嵌合方式,并有效连接于Cas9蛋白时,在II型特异性启动子的驱动下,Cas9蛋白能够被正常翻译且sgRNA能够被转录,两者相互作用,剪切靶点位置,即II型特异性启动子驱动下的Cas9/sgRNA(靶点和sgRNA插入到内含子内部的嵌合方式)系统能够正常行使剪切和/或编辑功能。综上所述,本发明将靶点和sgRNA与Cas9蛋白整合在一个表达盒中,并使用II型启动子驱动表达,不仅拓宽了sgRNA可利用的启动子范围,有利于实现特异组织的基因编辑,同时消除了对于靶点第一位碱基的限制,扩大了靶点选择范围,而且可以减少载体构建过程中表达盒的数量、简化载体,还能避免由于多个表达盒使用相同的启动子而发生重组导致部分元件的丢失的问题;本发明提供的由II型启动子驱动的基因编辑系统获得的编辑效率与常规体系相当,并且可以直接应用于实际生产中。最后所应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1