一种替诺福韦酯富马酸盐二聚体杂质的合成方法与流程
本发明涉及一种替诺福韦酯富马酸盐二聚体杂质的合成方法。
背景技术:
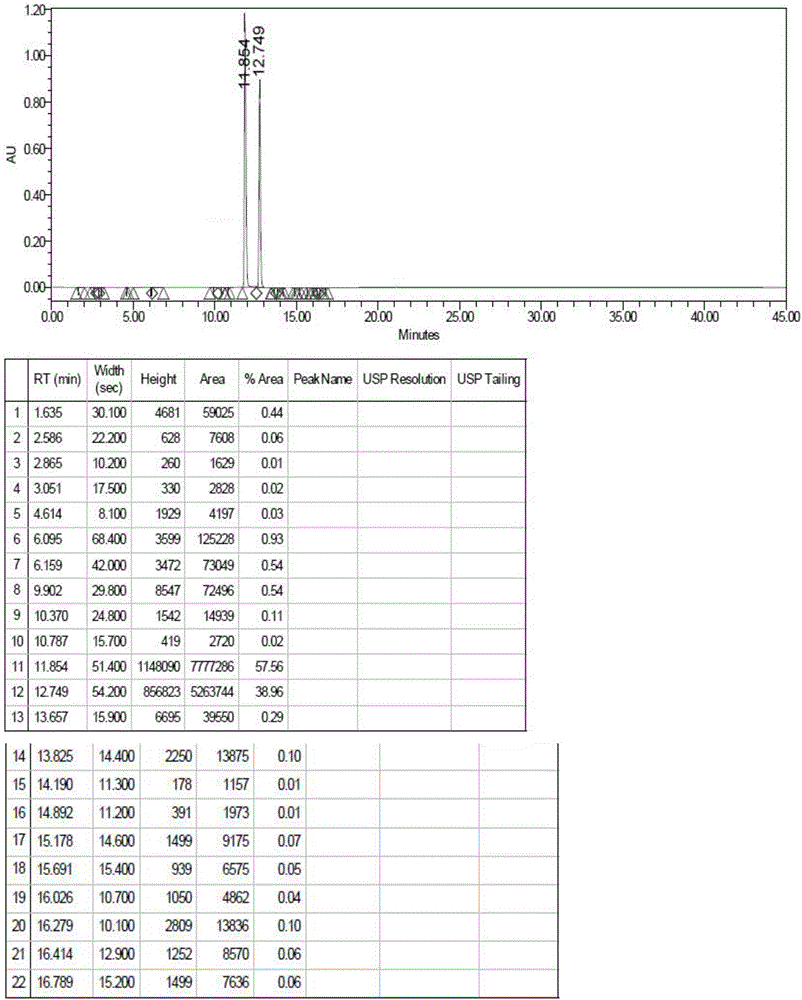
:替诺福韦酯富马酸盐由吉利德公司开发,商品名为Viread,于2001年10月在美国获批上市,用以治疗HIV、HBV感染,与其他逆转录酶拟制剂共同使用。该药物在2013年8月在中国获批上市,由葛兰素史克保留替诺福韦(商品名:韦瑞德)在中国上市的独有权,并负责该药用于治疗HBV感染患者的注册。由于具有很强的抗病毒活性,韦瑞德和恩替卡韦成为目前中国肝病防治指南推荐的抗乙肝病毒的一线用药。此外,由于治疗效果确切、适用性好、剂量合适,替诺福韦酯也是多个治疗指南推荐使用的一线抗HIV药物。替诺福韦酯及其复方制剂已成为目前销售额最大的抗艾滋病药物。据世界卫生组织统计,全世界有20亿人感染乙肝病毒,3.5亿以上的人患有慢性(长期)肝脏感染疾病,其中,中国的乙肝患者约占1/3,中国目前每年用于乙肝治疗的总费用超过1000亿,已成为全球最大的乙肝药物市场。而乙肝目前被认为还不能被彻底治愈,因此抗病毒被认为是其中最基本、最重要的治疗手段。替诺福韦酯富马酸盐杂质可以用于替诺福韦酯富马酸盐生产中的杂质定性及定量分析,从而可以提高替诺福韦酯富马酸盐的质量标准,为人民群众安全用药提供重要的指导意义。式Ⅰ为一种替诺福韦酯富马酸盐二聚体杂质的结构式:现有技术中,未见关于该杂质的合成方法介绍,这严重限制了人们对替诺福韦酯富马酸盐二聚体杂质的研究进程,因此,提供一种合理的替诺福韦酯富马酸盐二聚体杂质的合成方法,显得尤为必要。技术实现要素:本发明所要解决的技术问题是针对现有技术的现状,提供一种替诺福韦酯富马酸盐二聚体杂质的合成方法,该方法所得杂质的纯度大于90%,为替诺福韦酯富马酸盐生产中的杂质定性及定量分析提供了条件。本发明解决上述技术问题所采用的技术方案为:一种替诺福韦酯富马酸盐二聚体杂质的合成方法,其特征在于包括以下步骤:(1)将多聚甲醛加入酸试剂中,完全溶清后与下式Ⅱ化合物进行反应;(2)将步骤(1)所得溶液蒸除酸试剂至尽,得到下式Ⅲ化合物;(3)将步骤(2)所得式Ⅲ化合物、氯甲酸碳酸异丙酯、三乙胺、TBAB加入有机溶剂中反应,反应完毕后过滤,将所得滤液蒸干即得到目标产物替诺福韦酯富马酸盐二聚体杂质,即下式Ⅰ化合物。作为优选,步骤(1)中所述的酸试剂为盐酸或氢溴酸。在上述方案中,步骤(1)中多聚甲醛加入酸试剂中后在35~40℃下搅拌溶解,并与式Ⅱ化合物在10~15℃下进行反应。在上述各优选方案中,步骤(3)中的反应温度为50~60℃,反应时间为4~6h。优选地,步骤(3)反应体系中所使用的有机溶剂为N-甲基吡咯烷酮,DMF,DMSO中的一种。为了提高收率,本发明对步骤(3)所得滤饼进行了以下处理,步骤(3)的过滤温度为25~35℃,所得滤饼经有机溶剂淋洗得到有机层,对该有机层进一步洗涤、萃取、蒸干得到所述的目标产物替诺福韦酯富马酸盐二聚体杂质。优选地,用于淋洗滤饼的有机溶剂为乙酸乙酯、乙酸异丙酯中的一种。步骤(3)反应体系的有机溶剂采用强质子溶剂,对化合物III有很好的溶解度,便于反应顺利进行;淋洗滤饼采用的溶剂便于后处理中水洗及纯化操作,便于分离去除。与现有技术相比,本发明的优点在于:本发明提供了一种全新的替诺福韦酯富马酸盐二聚体杂质的合成方法,该方法在酸试剂和多聚甲醛存在下,以替诺福韦或替诺福韦一水合物为反应原料先制备出式Ⅲ化合物,然后使式Ⅲ化合物在氯甲基碳酸异丙酯及催化剂TBAB存在下制备得到目标产物化合物Ⅰ,整个合成方法路线短、操作步骤简单,所得产物的纯度在90%以上,为替诺福韦酯富马酸盐的杂质定性及定量分析奠定了基础。附图说明图1为本发明实施例1中式Ⅲ化合物的LC-MS图谱;图2为本发明实施例1中式Ⅲ化合物的HPLC图谱;图3为本发明实施例1中式Ⅰ化合物的核磁共振氢谱;图4为本发明实施例1中式Ⅰ化合物的LC-MS图谱;图5为本发明实施例1中式Ⅰ化合物的HPLC图谱。具体实施方式以下结合附图实施例对本发明作进一步详细描述。实施例1:本实施例中替诺福韦酯富马酸盐二聚体杂质的合成方法包括以下步骤:(1)室温下向250mL的四口烧瓶中加入多聚甲醛5g、体积浓度为15%的盐酸150mL,加毕,升温至35~40℃搅拌保温1h,完全溶清后降温至10~15℃并在该温度下加入10g下式Ⅱ化合物,同温下保温反应48h;(2)步骤(1)反应毕,减压蒸除盐酸至尽,得到式Ⅲ化合物;如图1可以看出,该步骤所得产物的分子量与目标产物式Ⅲ化合物一致;如图2所示,所得式Ⅲ化合物的纯度HPLC=57.56%;(3)将步骤(2)所得式Ⅲ化合物加入250mL的四口烧瓶中,并依次加入27g氯甲基碳酸异丙酯、30mLN-甲基吡咯烷酮、20mL三乙胺、5.3gTBAB,搅拌升温至50~60℃并在该温度下搅拌4h,然后降温至30~35℃过滤,收集所得滤液A;用100mL乙酸乙酯淋洗滤饼,抽至干,所得乙酸乙酯层中加入200mL饱和食盐水洗涤,静置分层,盐水层用40mL*3乙酸乙酯提取3次,合并每次提取的乙酸乙酯层并用50g无水硫酸钠干燥至澄清;过滤,用30mL乙酸乙酯淋洗无水硫酸钠,乙酸乙酯层去减压蒸馏,蒸除乙酸乙酯至尽所得液体与上述滤液A混合蒸干,所得产物采用DCM:MeOH=10:1过柱纯化得到目标产物式Ⅰ化合物0.51g。如图3所示,数据分析如下:质子编号化学位移(ppm)峰的多重性*质子数目18.36s2H27.92s2H37.26-7.19br2H45.63-5.55m10H54.91-4.83m4H64.31-4.28m2H74.13-4.08dd,J=14.0Hzand6.8Hz2H83.90-3.89m4H93.71-3.65dd,J=14.0Hzand8.8Hz2H101.27-1.24m24H111.10-1.08d,J=6.4Hz6H*s=单峰,d=二重峰,t=三重峰,q=四重峰,m=多重峰,dd=双二重峰,brs=宽峰、单峰如图4所示,质谱图中在m/z=526处的较强离子峰对应于样品的[1/2M+H]+离子,由此可知样品的分子量为1050.35,在m/z=1051处的较强离子峰对应于样品的[M+H]+离子,由此同样可知样品的分子量为1050.35。综合上述对图3、图4的分析结构可知,式Ⅰ化合物的结构式符合要求。如图5所示,所得式Ⅰ化合物的纯度HPLC=92.95%。实施例2:本实施例中替诺福韦酯富马酸盐二聚体杂质的合成方法包括以下步骤:(1)室温下向250mL的四口烧瓶中加入多聚甲醛5g、体积浓度为15%的氢溴酸150mL,加毕,升温至35~40℃搅拌保温1h,完全溶清后降温至10~15℃并在该温度下加入15g下式Ⅱ化合物,同温下保温反应48h;(2)步骤(1)反应毕,减压蒸除氢溴酸至尽,得到式Ⅲ化合物;(3)将步骤(2)所得式Ⅲ化合物加入250mL的四口烧瓶中,并依次加入27g氯甲基碳酸异丙酯、30mLDMSO、20mL三乙胺、5.3gTBAB,搅拌升温至50~60℃并在该温度下搅拌4h,然后降温至30~35℃过滤,收集所得滤液A;用100mL乙酸异丙酯淋洗滤饼,抽至干,所得乙酸异丙酯层中加入200mL饱和食盐水洗涤,静置分层,盐水层用40mL*3乙酸异丙酯提取3次,合并每次提取的乙酸异丙酯层并用50g无水硫酸钠干燥至澄清;过滤,用30mL乙酸异丙酯淋洗无水硫酸钠,乙酸异丙酯层去减压蒸馏,蒸除乙酸异丙酯至尽所得液体与上述滤液A混合蒸干,所得产物采用DCM:MeOH=10:1过柱纯化得到目标产物式Ⅰ化合物0.34g。实施例3:本实施例中替诺福韦酯富马酸盐二聚体杂质的合成方法包括以下步骤:(1)室温下向250mL的四口烧瓶中加入多聚甲醛5g、体积浓度为15%的盐酸150mL,加毕,升温至35~40℃搅拌保温1h,完全溶清后降温至10~15℃并在该温度下加入10g下式Ⅱ化合物,同温下保温反应48h;(2)步骤(1)反应毕,减压蒸除盐酸至尽,得到式Ⅲ化合物;如图1可以看出,该步骤所得产物的分子量与目标产物式Ⅲ化合物一致;(3)将步骤(2)所得式Ⅲ化合物加入250mL的四口烧瓶中,并依次加入27g氯甲基碳酸异丙酯、30mLDMF、20mL三乙胺、5.3gTBAB,搅拌升温至50~60℃并在该温度下搅拌4h,然后降温至30~35℃过滤,收集所得滤液A;用100mL乙酸乙酯淋洗滤饼,抽至干,所得乙酸乙酯层中加入200mL饱和食盐水洗涤,静置分层,盐水层用40mL*3乙酸乙酯提取3次,合并每次提取的乙酸乙酯层并用50g无水硫酸钠干燥至澄清;过滤,用30mL乙酸乙酯淋洗无水硫酸钠,乙酸乙酯层去减压蒸馏,蒸除乙酸乙酯至尽所得液体与上述滤液A混合蒸干,所得产物采用DCM:MeOH=10:1过柱纯化得到目标产物式Ⅰ化合物0.455g。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1