一种泊沙康唑及其中间体的制备新方法与流程
本发明属于药物技术领域,涉及泊沙康唑中间体的制备方法及其用于制备泊沙康唑的新方法。
背景技术:
泊沙康唑(posaconazole;cas登记号为171228-49-2),化学名为4-[4-[4-[4-[[(3r,5r)-5-(2,4-二氟苯基)-5-(1,2,4-三唑-1-基甲基)氧杂戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基]-2-[(2s,3s)-2-羟基戊-3-基]-1,2,4-三唑-3-酮。由schering-plough研发,2005年首次在德国上市,商品名为noxafil。随后在澳大利亚、英国、美国、欧盟等多个国家上市,用于预防和治疗侵袭性真菌感染、耐药或其他药物无效的侵袭性真菌感染。泊沙康唑为二代新型三唑类药物,结构式如下。
泊沙康唑是新型三唑类抗真菌药物,为伊曲康唑衍生物,难溶于水,其抗真菌活性优于其他所有三唑类药物。该类药物代谢稳定、低毒、生物利用度高、低蛋白结合率、分布广泛,具有较长的半衰期以及较好的耐受性和疗效。在酸性胃液环境下的生物利用度高,特别是进食脂肪食物时,较低剂量即可达到吸收饱和。与伊曲康唑不同,泊沙康唑更容易透过脑脊液屏障,脑中生物利用度较高。
已经公开报道的制备泊沙康唑的方法主要有以下几种。
在专利wo9517407、美国专利us5710154、us5714490、欧洲专利ep0736030及中国专利zl1142828中介绍了通过1-氯代-2,4-二氟苯乙酮经witting反应得到2-(2,4-二氟苯基)丙烯醇,再经过sharpless环氧化、亲核取代、缩合、还原等多步反应得到产物的方法。
该方法经过11步反应得到最终产品,反应步骤较长,专利中多步反应都提到采用柱层析的方式对中间体进行分离纯化,收率较低,造成生产成本较高,不利于工业生产。具体反应式如下。
在专利wo9633178中介绍了s-乳酸乙酯经吡咯烷酮保护,苄基保护羟基,然后再与哌嗪中间体iii和式vi化合物反应,最后采用钯碳和甲酸脱保护基、氨水水解合并反应,得到泊沙康唑的方法。
该方法反应路线过长,且用到强活泼性的格氏试剂和硼氢化锂,以及具有基因毒性的肼和酰氯,不利于工业生产。具体反应式如下。
专利wo2009141837公开了合成泊沙康唑及其中间体的方法,采用式iii化合物和式iv化合物反应得到式v化合物,然后通过钯碳加氢反应得到泊沙康唑。
该方法中的钯碳加氢反应需要特殊设备,工艺操作中容易出现燃烧、爆炸等危险。具体反应式如下。
中国专利cn102863431公开了一种合成泊沙康唑的方法,采用式(iii)化合物和式(iv)化合物反应,得到式(v)化合物用浓盐酸脱除保护基反应得到泊沙康唑(i)。
该方法采用的浓盐酸酸性较强,反应温度高,产品杂质增多;而且对设备和环境具有较大的腐蚀性和污染性。具体反应式如下。
鉴于泊沙康唑的药用价值,有必要寻找一种操作简便、收率良好、成本低廉,并且产品质量符合要求的合成方法。
技术实现要素:
本发明旨在寻找一种制备泊沙康唑及其中间体的新方法,根据绿色化学和经济合成理念,提供一种改进的化合物制备方法。该法条件温和,工艺简单,经济环保,利于工业化生产。
为实现上述目的,本发明提供了如下技术方案:一种泊沙康唑及中间体的新的制备方法。
所述制备方法主要包括以下步骤:
(1)以2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4[4-(4-羟基苯基-1-哌嗪基)苯基-3h-1,2,4-三氮唑-3-酮(iii)和(5r-cis)-甲苯-4-磺酸-5-(2,4-二氟苯基)-5-(1h-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(iv)为原料,在非质子性溶剂中,于碱性条件下,通过醚合成反应,得到式v化合物;
(2)在甲酸和钯碳作用下,式v化合物进行脱保护基和酯化反应生成式ii化合物;
(3)在水溶性溶剂中,式ii化合物在碱溶液条件下进行酯水解反应,生成泊沙康唑(式i)。
本发明人对中间体ii的制备方法进行了大量研究,通过钯碳催化氢化方法,采用式v化合物制备泊沙康唑,反应条件温和,无需高危险性的高温氢化反应,安全系数高;同时,不需要特殊要求的氢化设备和氢化车间,大大降低了生产成本。
作为本发明的进一步改进,为能有效提高终产品泊沙康唑的纯度和收率,发明人对中间体ii进行提取纯化,然后经氨水、三乙胺等弱碱处理后生成泊沙康唑(i)。所得泊沙康唑纯度高、收率高,所需重结晶纯化次数少,简化了生产工艺,利于工业化生产。
发明人在试验中发现,以甲醇、乙醇、异丙醇溶液为反应溶剂通过加水析晶制备泊沙康唑时,由于中间体ii和泊沙康唑快速析晶,晶体包裹中间体ii和其他中间产物等杂质,导致中间体ii反应不完全,且杂质增多。本发明采用三乙胺-甲醇作为泊沙康唑中间体ii酯水解的反应体系,巧妙地避免了上述缺陷,可以使反应充分进行,所得产品杂质更少,收率更高。
此外,采用乙醇、异丙醇作为反应溶剂,所需反应时间长,温度高。因此,采用本发明方法可有效降低工业化生产的成本。
本申请方法相对于原有技术公开的方法是一种显著的改进:
(1)就工艺的安全性方面,专利wo9633178方法中介绍了s-乳酸乙酯经吡咯烷酮保护,苄基保护羟基,然后再与哌嗪中间体和式vi化合物反应,得到泊沙康唑。该方法用到强活泼性、易燃易爆的格氏试剂和四氢铝锂,以及具有基因毒性的肼,毒性较大。本发明专利使用的溶剂均为常规溶剂,不涉及较大毒性的试剂。
专利wo2009141837方法采用式iii化合物和式iv化合物为原料合成式v化合物,式v化合物以氢气和钯碳催化氢化脱保护得到泊沙康唑。该方法使用氢气进行高温高压催化氢化反应,后者在工业上属于高度危险反应,存在燃烧爆炸的安全风险。本发明专利方法以式iii化合物和式iv化合物为原料合成式v化合物,式v化合物用钯碳和甲酸脱保护基、氨水处理得到泊沙康唑,工艺操作安全、简便,未用到高压、易爆等危险性大的工艺步骤。
(2)就工艺的产业化方面,专利wo9517407、美国专利us5710154、us5714490、欧洲专利ep0736030及中国专利zl94195025.5中介绍了通过1-氯代-2,4-二氟苯乙酮经witting反应得到2-(2,4-二氟苯基)丙烯醇,再经过sharpless环氧化、亲核取代、缩合、还原等多步反应得到产物。该方法经过11步反应最终得到产品,专利中多步反应都提到采用柱层析的方式对中间体进行分离纯化,不利于工业生产。本专利方法均采用常规反应条件,无需用到特殊设备,且整个过程操作简便,产品纯度高,工艺总收率大幅提高,给产业化带来极大便利。
(3)就泊沙康唑产品纯化和产品质量方面,专利wo9633178方法中,式v化合物脱保护基后继续和甲酸反应生成中间体(ii)泊沙康唑甲酸酯,直接进行氨水水解,不进行纯化,这会导致终产品杂质增多,产品颜色加深,纯化难度大,产品收率低;而且含重金属的钯碳很难通过过滤、析晶方法除去。本发明分别进行脱保护基、酯水解两步反应,将脱保护后生成的泊沙康唑甲酸酯进行纯化,得到中间体(ii),最后对中间体(ii)进行氨水水解得到终产品泊沙康唑(i),该方法终产品纯化难度极大降低,减少了产品重结晶次数,而且产品杂质明显减少,色泽更好,泊沙康唑粗品纯度hplc检测结果为99.5%以上,产品收率由60.1%大幅提高到86.6%,大大降低了生产成本。
附图简要说明
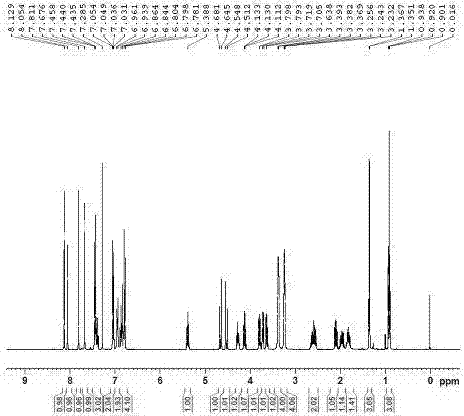
附图1实施例1所得式v化合物的1h-nmr图;
附图2实施例1所得式ii化合物的1h-nmr图;
附图3实施例1所得式ii化合物的13c-nmr图;
附图4实施例4所得泊沙康唑(i)的1h-nmr图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面,将对本发明的具体实施方式进行详细描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1:
步骤a.式v化合物的制备:
100ml三口瓶中加入2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4[4-(4-羟基苯基-1-哌嗪基)苯基-3h-1,2,4-三氮唑-3-酮(式iii)(6.08g)和dmso(30ml),搅拌溶解。加入25%氢氧化钠水溶液(1.7ml),混合物搅拌15min。加入(5r-cis)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1h-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(式iv)(6.12g),混合物在40-50℃搅拌反应12-18h。剧烈搅拌下将反应液倒入水(60ml)中,继续剧烈搅拌30min,抽滤,滤饼用水(30ml)洗涤,50℃真空干燥,得到式(v)化合物8.83g,收率95.6%。
lc-ms(esi):791(m+h);
1hnmr(400mhz,cdcl3)δppm:8.14(s,1h),7.82(s,1h),7.58(s,1h),7.44~7.40(m,3h),7.28~7.25(m,6h),7.04(d,j=9.2hz,2h),6.95(m,2h),6.88~6.79(m,4h),4.68~4.63(m,2h),4.54(d,j=14.4hz,1h),4.42(d,j=12hz,1h),4.24(m,1h),4.14(m,1h),3.82(m,2h),3.72(m,1h),3.62(m,1h),3.39(bs,4h),3.26(bs,4h),2.60(m,2h),2.13(m,1h),1.98(m,1h),1.83(m,1h),1.29(d,j=6.4hz,3h),0.91(t,j=7.2hz,3h)。
步骤b.式ii化合物的制备:
100ml三口瓶中加入式(v)化合物(8.81g)和甲酸(30ml),搅拌溶解。加入10%pd/c(0.88g),混合物在50-60℃下搅拌反应4-6h。冷却至室温,抽滤,滤液在30-40℃减压蒸干,加入甲醇45ml,滴加氨水调节ph值至7-8,析晶,抽滤,滤饼于40℃真空干燥4-6h,得到式(ii)化合物7.75g,收率95.5%。
lc-ms(esi):729(m+h);
1hnmr(400mhz,cdcl3)δppm:8.13(s,1h),8.05(s,1h),7.81(s,1h),7.68(s,1h),7.46~7.44(m,3h),7.05~7.03(m,2h),6.95(d,j=8.8hz,2h),6.86~6.78(m,4h),5.38(m,1h),4.66(d,j=14.4hz,1h),4.52(d,j=14.4hz,1h),4.30(m,1h),4.13(m,1h),3.79(m,1h),3.70(m,1h),3.64(m,1h),3.39(bs,4h),3.26(bs,4h),2.60(m,2h),2.13(m,1h),1.98(m,1h),1.83(m,1h),1.36(d,j=6.4hz,3h),0.92(t,j=7.2hz,3h)。
13cnmr(400mhz,cdcl3)δppm:164.09,163.97,161.49,160.27,160.18,157.84,157.73,153.07,151.10,150.63,144.57,134.52,128.64,125.45,123.45,118.56,116.66,115.20,111.23,104.63,84.05,70.89,69.00,59.90,55.98,50.69,49.17,38.87,37.49,22.50,17.30,10.39。
实施例2:
步骤a.式v化合物的制备:
100ml三口瓶中加入2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4[4-(4-羟基苯基-1-哌嗪基)苯基-3h-1,2,4-三氮唑-3-酮(式iii)(5.13g)和dmf(50ml),搅拌溶解。加入碳酸铯(6.51g),混合物搅拌15min。加入(5r-cis)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1h-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(式iv)(5.39g),混合物于60-70℃搅拌反应12-18h。剧烈搅拌下将反应液倒入水(100ml)中,继续剧烈搅拌30min,抽滤,滤饼用水(50ml)洗涤,50℃真空干燥,得到式(v)化合物7.46g,收率94.4%。
步骤b.式ii化合物的制备:
100ml三口瓶中加入式(v)化合物(7.42g)和甲酸(30ml),搅拌溶解。加入10%的pd/c(0.37g),在50-60℃下搅拌10-14h,抽滤,滤液于30-40℃减压浓缩,残留物加甲醇37ml溶解,加浓氨水调节ph值至7-8,析出固体,抽滤,滤饼于40℃真空干燥4-6h,得到式(ii)化合物6.56g,收率96.2%。
实施例3:
步骤a.式v化合物的制备:
100ml三口瓶中加入2-[(1s,2s)-1-乙基-2-苄氧基丙基]-2,4-二氢-4-[4[4-(4-羟基苯基-1-哌嗪基)苯基-3h-1,2,4-三氮唑-3-酮(式iii)(8.25g)和dmf(60ml),搅拌溶解。加入碳酸钾(5.01g),混合物搅拌15min。加入(5r-cis)-甲苯-4-磺酸5-(2,4-二氟苯基)-5-(1h-1,2,4-三氮唑-1-基)甲基四氢呋喃-3-基甲基酯(式iv)(8.32g),混合物于70-80℃搅拌14-18h。剧烈搅拌下将反应液倒入水(200ml)中,继续剧烈搅拌30min,抽滤,滤饼用水(100ml)洗涤,50℃真空干燥,得到式(v)化合物12.09g,收率95.2%。
步骤b.式ii化合物的制备:
100ml三口瓶中加入式(v)化合物(15.00g)和甲酸(60ml),搅拌溶解。加入10%的pd/c(0.50g),于50℃搅拌8-10h。抽滤,滤液在30-40℃减压浓缩,残留物加甲醇溶解,加三乙胺调节ph值至7-8,加水析晶,抽滤,滤饼于30-40℃真空干燥4-6h,得到式(ii)化合物8.84g,收率96.2%。
实施例4:泊沙康唑(i)的制备
将实施例1所得式ii化合物(7.23g)和甲醇(70ml)加至250ml三口瓶中,于60-70℃搅拌溶解,加入三乙胺(1.21g),继续搅拌1h,冷却至室温,抽滤,滤饼用50%甲醇水溶液(10ml)淋洗,于50℃真空干燥,得到泊沙康唑(i)6.93g,收率95.8%,hplc检测纯度99.94%。
lc-ms(esi):701(m+h);
1hnmr(400mhz,cdcl3)δppm:8.14(s,1h),7.82(s,1h),7.69(s,1h),7.47~7.45(m,3h),7.07~7.05(m,2h),6.96(bs,2h),6.87~6.80(m,4h),4.67(d,j=14.4hz,1h),4.53(d,j=14.4hz,1h),4.14~4.03(m,3h),3.80(m,1h),3.71(m,1h),3.64(m,1h),3.41(bs,4h),3.27(bs,4h),3.10(d,1h),2.60(m,2h),2.11(m,1h),2.03(m,1h),1.92(m,1h),1.62(bs,1h),1.24(d,j=6hz,3h),0.97(t,3h)。
实施例5:泊沙康唑(i)的制备
将实施例2所得式(ii)化合物(6.51g)和甲醇(65ml),加至100ml三口瓶中,于60-70℃搅拌溶解,加入氨水(2.13g),继续搅拌2h,加入水65ml,冷却至室温,抽滤,滤饼用50%甲醇水溶液(10ml)淋洗,于50℃真空干燥,得到泊沙康唑(i)6.21g,收率95.5%,hplc检测纯度99.89%。
实施例6:泊沙康唑(i)的制备
将实施例3所得式ii化合物(4.32g)和乙醇(43ml)加至100ml三口瓶中,于70-80℃搅拌溶解,加入三乙胺(0.73ml),继续搅拌3h,滴加水43ml,冷却至室温,抽滤,产品用50%乙醇水溶液(10ml)淋洗,于50℃真空干燥,得到泊沙康唑(i)4.12g,收率95.3%,hplc检测纯度99.91%。
实施例7:泊沙康唑(i)的制备
将实施例4所得式ii化合物(4.27g)和异丙醇(43ml)加至100ml三口瓶中,于80-90℃搅拌溶解,加入三乙胺(0.72ml),继续搅拌4h,冷却至室温,滴加水43ml,抽滤,滤饼用50%乙醇水溶液(10ml)淋洗,于50℃真空干燥,得到泊沙康唑(i)4.04g,收率94.8%。
以上所述具体实施方式仅用于具体说明本发明的精神,本发明的保护范围并不局限于此,对于本技术领域的技术人员来说,当然可根据本说明书中所公开的技术内容,通过变更、置换或变型的方式轻易做出其它的实施方式,这些其它的实施方式都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!