用于表征基于时间的肝毒性的方法与流程
简介
新药开发中的一个主要障碍是推进能够展示足以获得监管部门批准的效力和安全性的药物候选。有前景的候选经常在临床前或临床阶段失败。尽管存在许多原因使药物候选在开发过程中流失,一个最常见的原因涉及潜在肝脏毒性的安全相关发现。本领域需要减少由于发现肝脏毒性导致临床和后期临床前失败而产生的开销、减少鉴别安全相关问题的时间和加速通过基础发现研究鉴别的候选化合物的决策。
目前采用的众多体外研究支持以最高成功概率选择候选,但在检测肝毒性上存在缺陷。肝毒性可由一种或多种机制导致,包括基因毒性(诱变、诱裂)、cyp抑制(包括时间抑制)、cyp诱导、g-sh加合物形成、与蛋白的共价结合和herg电流抑制、脂肪变性、肉芽肿、形成反应性代谢物和胆汁淤积。使用常规肝细胞系统的体外细胞毒性评估甚至不足以充分预测体内肝毒性可能性,其包括提供短期或急性测试的基于细胞的体外模型,经常使用温育,获得仅24小时或更短的短期时间点的数据,或仅获得在一个该时间点的数据。事实上,相关领域的众多毒理学家认为源自如此短期、“急性”体外测试的数据在临床前决策中具有很少或没有可操作的价值,且经常将药物化合物继续推进入随后(且越来越昂贵)的开发阶段,尽管这些模型可生成阳性肝毒性响应的发现(有时在本领域称为“肝脏信号”)。因此,本领域需要表征肝毒性的新型和改进的方法和系统。本发明满足了这些和其他需求。
概述
目前体外系统局限性的原因之一在于其一般因为有限和/或递减的代谢能力而限于24小时的读取。本发明利用在培养中能够在体外维持功能性达延长时间的肝细胞培养物,所述时间可达例如14、21或28小时。部分基于使用该系统,本发明提供包括在1、2、3、4、5、6、7或更多天,或14天或更长时间的多个时间点测量培养物中肝毒性的方法。在一些实施方案中,提供的方法允许以相对使用本领域的方法所实现的有所改进的方式预测测试化合物的肝毒性潜力。在一些实施方案中,所述方法可用于量化时间上不同、连续的点生成的毒性的测量和预估间的关系。通过比较在24小时测量的测试化合物的毒性(其中毒性在第7天或在第14天测量),发现了毒性随时间的变化提供了非常有意义的工具来在体内评估测试化合物在体内的肝毒性。
因此,在一些实施方案中,本发明提供表征测试化合物基于时间的肝毒性的方法。在一些实施方案中,所述方法包括a)将包含肝细胞的第一体外培养物与测试化合物温育第一培养时间;b)测量所述测试化合物在所述第一培养时间内对第一体外培养物的肝细胞的至少一种细胞毒性效应以由此定义所述测试化合物在第一培养时间内的肝毒性;c)将包含肝细胞的第二体外培养物与测试化合物温育长于第一培养时间的第二培养时间;d)测量所述测试化合物在第二培养时间内对第二体外培养物的肝细胞的至少一种细胞毒性效应以由此定义所述测试化合物在第二培养时间内的肝毒性;和e)将所述测试化合物在第一培养时间内的肝毒性与所述测试化合物在第二培养时间内的肝毒性比较以由此表征所述测试化合物基于时间的肝毒性。在一些实施方案中,所述测试化合物在第二培养时间内的肝毒性大于所述测试化合物在第一培养时间内的肝毒性且所述测试化合物鉴别为展示基于时间的肝毒性。在一些实施方案中,所述测试化合物在第二培养时间内的肝毒性比所述测试化合物在第一培养时间内的肝毒性大至少预定义的阈值且所述测试化合物鉴别为展示基于时间的肝毒性。在一些实施方案中,所述测试化合物在第二培养时间内的肝毒性未比所述测试化合物在第一培养时间内的肝毒性大至少预定义的阈值且所述测试化合物鉴别为不展示基于时间的肝毒性。在一些实施方案中预定义的阈值选自约2、约3、约4、约5、约6、约7、约8、约9或约10。在一些实施方案中,预定义的阈值选自2、3、4、5、6、7、8、9或10。在一些实施方案中,预定义的阈值为2至5、3至5、2至4、2至6、4至8或5至10。
在一些实施方案中,第一和第二体外培养物包含分离的肝细胞。在一些实施方案中,所述分离的肝细胞基本上分散在固体基质的表面上。在一些实施方案中,所述肝细胞为原代肝细胞。在一些实施方案中,所述肝细胞为干细胞衍生的。
在一些实施方案中,第一和第二体外培养物进一步包含至少一种限定的基质细胞类型。
在一些实施方案中,第一和第二体外培养物包含至少一种以下述微型模式构造的培养的肝细胞:含有肝细胞和至少一种其他细胞类型并粘附于基质材料的微型模式;肝细胞以细胞的球形构造成簇的微型模式,其封装或未封装于培养基液滴中或粘附或未粘附于微珠;肝细胞贮存在“三明治”培养物中的微型模式,其中肝细胞稳定于基底和胶原蛋白或其他凝胶或蛋白质的覆盖层间;和肝细胞嵌入膜、晶格、支架或天然存在或人工材料中或在其上的微型模式,所述人工材料包括但不限于含有源自海藻酸盐的材料的支架或晶格、人工半透膜或含有培养的毛细血管的床体。
在一些实施方案中,第一培养时间为12小时至2天。在一些实施方案中,第一培养时间是1天。在一些实施方案中,第二培养时间为3至28天。在一些实施方案中,第二培养时间为7至14天。在一些实施方案中,第一培养时间是1天且所述第二培养时间为7天或14天。
在一些实施方案中,“x”天的培养时间为至少“x”天的培养时间。在一些实施方案中,“x”天的培养时间为至少约“x”天的培养时间。在一些实施方案中,“x”天的培养时间为约“x”天的培养时间。
在一些实施方案中,所述方法进一步包括至少一个第三培养时间,并a)将包含肝细胞的第三体外培养物与测试化合物温育第三培养时间,所述第三培养时间比第一和第二培养时间更长;b)测量所述测试化合物在所述第三培养时间内对第三体外培养物的肝细胞的至少一种细胞毒性效应以由此定义所述测试化合物在第三培养时间内的肝毒性;和c)将第一、第二或第一和第二培养时间内测试化合物的肝毒性与在第三培养时间内测试化合物的肝毒性进行比较以由此表征测试化合物基于时间的肝毒性。
在所述方法的一些实施方案中,比较包括实施数学运算,其包括确定两个或多个测量量之间的算术比值,或利用积分或微积分的方法或利用概率、随机或模拟形式的测量或计算模型确定两侧测量的量之间的关系。
在一些实施方案中,步骤a)包括将包含肝细胞的多种第一体外培养物与所述测试化合物温育第一培养时间。
在一些实施方案中,步骤c)包括将包含肝细胞的多种第二体外培养物与所述测试化合物温育第二培养时间。
在一些实施方案中,步骤b)包括确定所述化合物在第一培养时间内的tc50。
在一些实施方案中,步骤d)包括确定所述化合物在第二培养时间内的tc50。
在一些实施方案中,步骤b)包括确定所述化合物在第一培养时间内的tc50,其中步骤d)包括确定所述化合物在第二培养时间内的tc50,且其中步骤e)包括确定所述化合物在第一培养时间内的tc50与所述化合物在第二培养时间内的tc50的比率以定义所述测试化合物基于时间的毒性评分。
附图简述
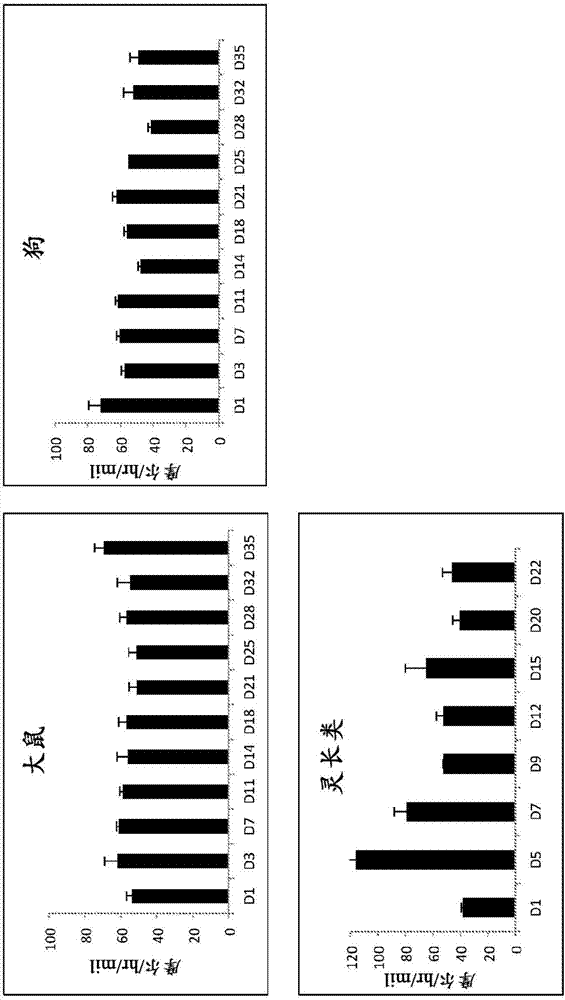
图1显示在本发明的大鼠、狗和灵长类肝细胞-基质细胞共培养物中i期酶功能是持久的。在所示时间点测量了7-羟基香豆素形成的速率。不同模式对于不同物种是明显的;然而,在每种情况中,i期酶都是功能持久的。自肝细胞接种日,大鼠和狗具有至少35天的i期活性且灵长类具有至少22天的i期活性。
图2显示在本发明的大鼠、狗和灵长类肝细胞-基质细胞共培养物中ii期酶功能是持久的。在所示时间点测量了7-羟基香豆素葡萄糖醛酸的形成速率。不同模式对于不同物种是明显的;然而,在每种情况中,ii期酶都是功能持久的。自肝细胞接种日,大鼠和狗具有至少35天的ii期活性且灵长类具有至少22天的ii期活性。
图3显示从三个不同批次的原代人肝细胞生成的肝细胞-基质细胞共培养物中持久的cyp3a4功能。cyp3a4的功能通过测量1-羟基咪达唑仑的形成速率分析。观察了基线中的一些批次比彼此差异,但自细胞接种日,全部批次都具有达35天的3a4活性。
图4显示从三个不同批次的原代人肝细胞生成的肝细胞-基质细胞共培养物中持久的cyp2c9功能。cyp2c9的功能通过测量4-羟基甲苯磺丁脲的形成速率分析。观察了基线中的一些批次比彼此差异,但自细胞接种日,全部批次都具有达35天的2c9活性。
图5显示使用人、狗、大鼠和灵长类肝细胞建立的肝细胞-基质细胞共培养物中的小管染色。
图6显示培养物中从第7天至第25天人肝细胞-基质细胞共培养物中肝细胞的cyp3a4活性是稳定的。到第7天,培养的灵长类肝细胞具有完成的重塑和稳定化的功能。至第25天功能保持稳定。稳定功能的窗口因此持续18天。值得注意的是两周的稳定培养对于大多数dmpk和tox应用是足够长的时间。
图7显示在人肝细胞-基质细胞共培养物中,在大多数或全部三个不同批次的人细胞中,培养的第7天至第18天间四种不同的cyp酶保持稳定。
图8显示对于i期代谢的多物种稳定性窗口。
图9显示对于i期代谢的多物种稳定性窗口。
图10比较了hepg2细胞系和肝细胞-基质细胞共培养物。氟他胺和酮康唑是肝毒性的。数据显示毒性母体化合物(氟他胺和酮康唑)的更多肝清除需要更高的母体浓度以产生等同的毒性水平。曲格列酮肝毒性由其肝毒性代谢物调节。曲格列酮的初级反应代谢物已确认为邻醌甲基化物(o-quinonemethide)。该代谢物一般由agsh偶联经由取代的苯并二氢吡喃环体系的氧化成为反应性邻醌甲基化物衍生物形成。数据显示毒性代谢物的更多生成需要较低的母体浓度以产生等同水平的毒性。总之,这些数据证明本发明的肝细胞-基质细胞共培养物相比hepg2单培养物的出色的预测能力。
图11显示多剂量的环磷酰胺对包含人、狗、灵长类或大鼠的原代肝细胞的肝细胞-基质细胞共培养物的肝毒性效应。使用celltiter-blue(promega)测定分析了细胞活力,n=4。数据显示延长给药持续时间增加环磷酰胺的细胞毒性。
图12显示处理6天后(三次给药),多剂量曲格列酮对包含人、狗、灵长类或大鼠的原代肝细胞的肝细胞-基质细胞共培养物的肝毒性效应(计算的tc50)。在图12所示的三个图中分别单独比较了狗、猴和大鼠与人的结果。
图13总结了环磷酰胺和曲格列酮的结果。这些为在4个不同物种中6天反复化合物暴露后生成的浓度依赖毒性曲线。自第0天,每种化合物每2天给药。在第6天,使用promegacelltiterblue测定测量刃天青到荧光试卤灵的转化。线粒体中试卤灵有效减少,因此该测定测量线粒体代谢活性。随后如下将数据用于生成tc50值(即响应施用的剂量在培养物中50%的细胞死亡的化合物的浓度)。对于曲格列酮:lc50=308μm(人)、lc50=267μm(狗)、lc50=207μm(猴)和lc50=317μm(大鼠)。对于环磷酰胺:lc50=5,523μm(人)、lc50=7,541μm(狗)、lc50=1,904μm(猴)、lc50=1,506μm(大鼠)。
图14给出了gsh和promegacelltiterblue测定的比较。在50*cmax完成测定。非常低的批次间差异性是本发明的系统和方法有效性和能力的进一步证明。
图15列举了在实施例中使用的20中模型肝毒性化合物。第三列表示化合物是否为已知的强肝毒素、已知的中度肝毒素或已知为无肝毒性。第二列表示由已知肝毒素诱导的肝毒性的大概或可能的机制。
图16给出了在24小时人肝细胞单培养物中和在24小时、7天和14天人肝细胞-基质细胞共培养物中观察到的20种化合物中的每一种肝毒性。tc50值的单位为微摩尔。
图17给出了在人肝细胞共培养物中观察到的基于时间的毒性信号的计算。
图18给出了在24小时大鼠肝细胞单培养物中和在24小时、7天和14天大鼠肝细胞-基质细胞共培养物中观察到的20种化合物中的每一种肝毒性。tc50值的单位为微摩尔。
图19给出了在大鼠肝细胞共培养物中观察到的基于时间的毒性信号的计算。
图20a、20b和20c是单个表的部分且显示基于时间的毒性信号与已知的测试化合物的毒性机制相关。
图21显示开发至临床前阶段的一组十种化合物基于时间的毒性信号与观察到的毒性相关。
图22显示开发至临床前阶段的一组十种化合物基于时间的毒性信号与观察到的毒性相关。
发明详述
在公开和描述本系统和方法以及其他实施方案之前,应该理解的是,本文所使用的术语仅用于描述特定实施方案的目的,而不是限制性的。必须注意,如说明书和所附权利要求书中所使用的,除非上下文另有明确说明,否则单数形式“一”,“一个”和“所述”包括复数指代物。
本文使用的术语“包含”与“包括”或“含有”是同义的,并且是包含性的或开放式的,并且不排除另外的未列举的成员,要素或方法步骤。
如本文所用,“肝毒性”可包括任何形式的化学驱动的肝损伤,包括但不限于通过选自下述的一种或多种机制导致的肝损伤:基因毒性(诱变、诱裂)、cyp抑制(包括时间抑制)、cyp诱导、g-sh加合物形成、与蛋白的共价结合和herg电流抑制、脂肪变性、肉芽肿、形成反应性代谢物和胆汁淤积。
a.肝细胞培养物
本公开提供了评估测试化合物的毒性的系统和方法,其包括将测试化合物施用于培养的肝细胞。在一些实施方案中,肝细胞以包含下述的微型模式构造:含有肝细胞和至少一种其他细胞类型并粘附于基质材料的微型模式;肝细胞以细胞的球形构造成簇的微型模式,其封装或未封装于培养基液滴中或粘附或未粘附于微珠;肝细胞贮存在“三明治”培养物中的微型模式,其中肝细胞稳定于基底和胶原蛋白或其他凝胶或蛋白质的覆盖层间;和肝细胞嵌入膜、晶格、支架或天然存在或人工材料中或在其上的微型模式,所述人工材料包括但不限于含有源自海藻酸盐的材料的支架或晶格、人工半透膜或含有培养的毛细血管的床体。在一些实施方案中,该系统和方法旨在通过将测定化合物施用于从心脏、肝脏、脑、中枢神经系统、肌肉、骨或骨髓、肺、胃肠壁、血脑屏障、角膜、男性或女性生殖器官、免疫系统或机体的任何其它器官等获取的培养细胞来评估测试化合物的毒性。
在一些实施方案中,培养的肝细胞以包含肝细胞和至少一种其他类型细胞的细胞的细胞共培养物的形式存在。在一些实施方案中,培养的肝细胞是至少包含肝细胞和共培养中表现出基质特征的细胞(基质细胞)的细胞共培养物的形式。
在一些实施方案中,肝细胞-基质细胞共培养物包含肝细胞和置于固体基质表面上的至少一种基质细胞类型。在一些实施方案中,将肝细胞-基质细胞培养物以故意的微型模式构造置于固体基质的表面上。在一些实施方案中,肝细胞-基质细胞培养物以其中两种类型的细胞相互散布或彼此混合的构造置于固体基质的表面上。在一些实施方案中,肝细胞-基质细胞共培养物包含肝细胞和至少一种基质细胞类型,它们一起展示球形构造。在一些实施方案中,肝细胞-基质细胞共培养物包含肝细胞和置于固体晶格内的至少一种基质细胞类型,其使得细胞展示和/或保持其共培养构造。在肝细胞-基质细胞培养物的一些实施方案中,肝细胞和单个基质细胞类型共同占共培养物中至少约95%、至少约96%、至少约97%、至少约98%、至少约99%、至少约99.5%、至少约99.9%或至少约99.99%的细胞。在肝细胞-基质细胞培养物的一些实施方案中,肝细胞和单个基质细胞类型共同占少于共培养物中细胞总数的任何前述百分比。
通常,肝细胞和基质细胞以1:10至10:1的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以2:10至10:2的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以2:10至4:10的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以4:10至6:10的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以6:10至8:10的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以8:10至1:1的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以1:1至10:8的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以10:8至10:6的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以10:6至10:4的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以10:4至10:2的比例存在于共培养物中。在一些实施方案中,肝细胞和基质细胞以约10:1、10:2、10:3、10:4、10:5、10:6、10:7、10:8、10:9、1:1、9:10、8:10、7:10、6:10、5:10、4:10、3:10、2:10或1:10的比例存在于共培养物中。
在一些实施方案中,肝细胞-基质细胞共培养物包含至少两种基质细胞类型。在一些实施方案中,肝细胞-基质细胞共培养物包含两种基质细胞类型,其在共培养物中各占至少约0.01%、至少约0.1%、至少约0.5%、至少约1%、至少约2%、至少约3%、至少约4%、至少约5%约10%、至少约20%、至少约30%或至少约40%的细胞。在一些实施方案中,肝细胞-基质细胞共培养物包含两种基质细胞类型,其在共培养物中各占总细胞数材料上不同的百分比。一些实施方案包含多于两组不同的基质细胞类型。在一些实施方案中,所有基质细胞类型在共培养中的功能贡献主要是基质性质的。在一些实施方案中,与至少一种第二基质细胞类型相比,至少一种基质细胞类型对细胞培养物提供更大的基质贡献。在一些实施方案中,至少一种第二基质细胞类型对本质上不是基质的细胞共培养物的整体功能或能力有贡献。
在肝细胞-基质细胞共培养物中的肝细胞可以是任何类型的肝细胞,其包括但不限于原代肝细胞、肝细胞系和通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞。在一些实施方案中,在肝细胞-基质细胞共培养物中的所有肝细胞都是原代肝细胞。在一些实施方案中,在肝细胞-基质细胞共培养物中的所有肝细胞来自至少一种肝细胞系。在一些实施方案中,肝细胞-基质细胞共培养物中的所有肝细胞为通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为包含原代肝细胞和至少一种肝细胞系的肝细胞的混合物。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为包含至少一种肝细胞系和通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞的混合物。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为包含原代肝细胞和通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞的混合物。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为包含原代肝细胞、至少一种肝细胞系和通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞的肝细胞的混合物。
在一些实施方案中,肝细胞-基质细胞共培养物包含来自单个供体哺乳动物的肝细胞。因此,在一些实施方案中,肝细胞-基质细胞共培养物包含全部获得自单个供体哺乳动物的原代肝细胞。在一些实施方案中,肝细胞-基质细胞共培养物包含来自多个肝细胞系的肝细胞,其中所述多个肝细胞系全部获得自单个供体哺乳动物的细胞。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞,其全部从单个供体哺乳动物生产。
在一些实施方案中,肝细胞-基质细胞共培养物包含来自多个供体哺乳动物的肝细胞。因此,在一些实施方案中,肝细胞-基质细胞共培养物包含获得自多个供体哺乳动物的原代肝细胞。在一些实施方案中,原代肝细胞为获得自相同物种的2、3、4、5、6、7、8、9或10个不同供体哺乳动物批次的混合物。在一些实施方案中,原代肝细胞为获得自多于一个物种的2、3、4、5、6、7、8、9或10个不同供体哺乳动物批次的混合物。在一些实施方案中,肝细胞-基质细胞共培养物包含肝细胞来自多个肝细胞系的肝细胞,其中所述多个肝细胞系获得自多个供体哺乳动物的细胞。在一些实施方案中,肝细胞系包含获得自2、3、4、5、6、7、8、9或10个不同供体哺乳动物的细胞系。在一些实施方案中,肝细胞-基质细胞共培养物中的肝细胞为通过干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)分化形成的肝细胞,其从多个供体哺乳动物生产。在一些实施方案中,肝细胞系包含通过来自2、3、4、5、6、7、8、9或10个不同供体哺乳动物的干细胞分化形成的肝细胞。
肝细胞可以是任何哺乳动物的肝细胞。在一些实施方案中,肝细胞是选自以下的哺乳动物的肝细胞:人、非人灵长类动物(如食蟹猴)、农场动物(如猪、马、牛和绵羊)、家养哺乳动物(如狗、猫、豚鼠、小猪和兔)和啮齿动物(如小鼠和大鼠)。
在优选的实施方案中,肝细胞为原代肝细胞。原代肝细胞可(但不必须)以冷冻保存的形式提供。冷冻保存的人肝细胞可以从商业企业例如thermofisher获得。冷冻保存的非人原代肝细胞可以从商业企业例如thermofisher(之前为lifetechnologies公司)、和bioreclamationivt其他等获得。冷冻保存的狗肝细胞可以从bioreclamationivt获得。冷冻保存的大鼠肝细胞可以从商业企业例如thermofisher(之前为lifetechnologies公司)、和bioreclamationivt其他等获得。在一些实施方案中,基质细胞类型来自与肝细胞相同类型的哺乳动物。在一些实施方案中,基质细胞类型来自与肝细胞不同类型的哺乳动物。
在一些实施方案中,肝细胞-基质细胞共培养物包含第三细胞类型。在一些实施方案中,第三细胞类型是基质细胞。在一些实施方案中,第三细胞培训并非基质细胞。在一些实施方案中,第三细胞类型是实质细胞。在一些实施方案中,第三细胞类型并非是实质细胞。在一些实施方案中,第三细胞类型选自ito细胞、内皮细胞、胆管细胞、免疫介导细胞和干细胞。在一些实施方案中,免疫介导性细胞选自巨噬细胞、t细胞、嗜中性粒细胞、树突细胞、肥大细胞、嗜酸性粒细胞和嗜碱性粒细胞。
在一些实施方案中,第三种细胞类型是库普弗细胞。在一些实施方案中,库普弗细胞占共培养物中细胞的至少约0.01%、至少约0.1%、至少约0.5%、至少约1%、至少约2%、至少约3%、至少约4%、至少约5%、至少约10%或更多。
在一些实施方案中,基质细胞类型是内皮细胞。在一些实施方案中,基质细胞类型是成纤维细胞。在一些实施方案中,基质细胞是原代细胞。在一些实施方案中,基质细胞获得自细胞系。在一些实施方案中,基质细胞是转化的细胞。在一些实施方案中,基质细胞从干细胞(如胚胎干细胞、成体干细胞和/或诱导的多能干细胞)体外分化。多种基质细胞来源如成纤维细胞为本领域已知可在肝细胞-基质细胞共培养物中利用。一个实例是nih3t3-j2细胞系(参见例如us2013/0266939a1)。
本领域教导了通过将肝细胞和基质细胞置于固体基质上以使肝细胞在第一步中以细胞岛构造附着于基质而改善培养中肝细胞功能的一些方面。(参见us2013/0266939a1)。具体而言,这样的方法依赖于在底物上形成肝细胞的细胞岛,肝细胞岛由非实质细胞类型如基质细胞类型包围。通过首先将细胞外基质组分或衍生物以岛状模式置于固体基质上,然后使肝细胞粘附于细胞外基质组分或衍生物而形成肝细胞岛。然后添加非实质细胞类型并允许“填充”不含肝细胞的基质部分。这种系统的基本特征是肝细胞不分散在基质表面。
在本发明的肝细胞-基质细胞共培养物中的一些实施方案中,肝细胞以如us2013/0266939a1中所述的细胞岛构造分布。然而,在优选的实施方案中,肝细胞基本上分散并与基质细胞混合穿过固体基质的表面。在一些优选的实施方案中,肝细胞基本上分散并与基质混合穿过固体基质的表面,并且肝细胞和基质细胞自身重组,使得它们的构造包含至少一些肝细胞被部分或完全构造在至少一些基质细胞上,和/或一些基质细胞部分或完全构造在至少一些肝细胞上。
如本文所用,关于肝细胞-基质细胞共培养物中肝细胞在固体支持物上的排列的“分散在表面上”是指适用于共培养物的至少一项以下标准:1)至少所述固体基质表面的约20%、至少约30%、至少约40%或至少约50%被至少一个肝细胞覆盖;2)共培养物中肝细胞的至少约2%,至少约5%或至少约10%位于与固体基质接触的基质细胞上;和3)通过将肝细胞加入包含至少一种细胞外基质组分的岛的固体基质中,肝细胞不被接种到固体基质上,以产生附着于固体基质的肝细胞岛。注意单个肝细胞可以被计数为符合标准1和标准2。
在优选的实施方案中,肝细胞-基质细胞共培养物的代谢功能是持久的。在一些实施方案中,培养持续的时间至少为一天,至少两天,至少三天,至少五天,至少七天,至少十天,至少十四天,至少二十一天,至少二十八天,至少三十五天,至少四十二天或超过至少四十二天。在一些实施方案中,肝细胞-基质细胞共培养物的功能通过测量肝细胞在共培养物中的基因表达、细胞功能、代谢活性、形态学和/或其组合的活性来确定。在一些实施方案中,通过测量至少一种cyp450酶的表达和/或活性水平来确定肝细胞-基质细胞共培养物的功能。至少一种cyp450酶的代谢表达和/或代谢活性的水平可以通过测量cyp450酶mrna的表达,通过测量cyp450酶蛋白的表达或通过cyp450酶活性的功能测定来测量。在一些实施方案中,代谢活性是所谓的i期代谢酶活性,例如cyp450酶活性。在一些实施方案中,cyp450酶是选自cyp1a2,cyp1b1,cyp2a6,cyp2b6,cyp2c,cyp2d6,cyp2e1,cyp2f1,cyp2j2,cyp3a4,cyp4a和cyp4b的cyp450酶。在一些实施方案中,i期代谢酶活性是非cyp酶活性,例如mao活性。在一些实施方案中,代谢活性是所谓的ii期代谢酶活性,例如ugt或sult酶活性。
如果肝细胞-基质细胞中共培养物的代谢功能比对照肝细胞单培养物的代谢功能更持久,则将肝细胞-基质细胞共培养物的代谢功能视为持久。在一些实施方案中,共培养物的代谢功能至少持续三天。在一些实施方案中,共培养物的代谢功能至少持续四天。在一些实施方案中,共培养物的代谢功能至少持续七天。在一些实施方案中,共培养物的代谢功能至少持续十四天。在一些实施方案中,共培养物的代谢功能持续至少二十一天。在一些实施方案中,共培养物的代谢功能至少持续二十八天。在一些实施方案中,共培养物的代谢功能至少需要三十五天。在一些实施方案中,共培养物的代谢功能至少持续四十二天。在一些实施方案中,共培养物的代谢功能至少持续四十二天以上。
在一些实施方案中,共培养物在无血清培养基或基本无血清培养基中培养。在一些实施方案中,共培养物在含有血清的培养基中培养。在一些实施方案中,培养基包含约0.1%的血清、约0.2%的血清、约0.3%的血清、约0.4%的血清、约0.5%的血清、约0.6%的血清、约0.7%的血清、约0.8%的血清、约0.9%的血清、约1%的血清、约2%的血清、约3%的血清、约4%的血清、约5%的血清、约6%的血清、约7%的血清、约8%的血清、约9%的血清或约10%的血清。在一些实施方案中,培养基包含至少约0.1%的血清、至少约0.2%的血清、至少约0.3%的血清、至少约0.4%的血清、至少约0.5%的血清、至少约0.6%的血清、至少约0.7%的血清、至少约0.8%的血清、至少约0.9%的血清、至少约1%的血清、至少约2%的血清、至少约3%的血清、至少约4%的血清、至少约5%的血清、至少约6%的血清、至少约7%的血清、至少约8%的血清、至少约9%的血清或至少约10%的血清。在一些实施方案中,培养基包含少于或等于约0.1%的血清、少于或等于约0.2%的血清、少于或等于约0.3%的血清、少于或等于约0.4%的血清、少于或等于约0.5%的血清、小于或等于约0.6%的血清、小于或等于约0.7%的血清、小于或等于约0.8%的血清、小于或等于约0.9%的血清、小于或等于约1%的血清、小于或等于约2%的血清、小于或等于约3%的血清、小于或等于约4%的血清、小于或等于约5%的血清、小于或等于约6%的血清、小于或等于约7%的血清、小于或等于约8%的血清、小于或等于约9%的血清或小于或等于约10%的血清。
在本公开的一些实施方案中,系统和方法提供了来自四种不同哺乳动物物种中的至少三种的可比较的肝细胞-基质细胞共培养物。
实施例中提供的实施方案利用某些原代肝细胞-基质细胞共培养物。然而,本发明不应被理解为限于细胞培养物的构造。本发明的全部范围包括任何在延长的培养时间内使细胞(例如肝细胞)维持在高代谢功能状态的体外细胞模型系统,使得测试化合物可以与细胞培养物在第一和第二培养时间温育,以便比较第一和第二培养时间内的毒性。
在一些实施方案中,不同的细胞类型代替肝细胞。例如,本发明包括但不限于测量来自除肝以外的器官(例如心脏和肾脏,作为两个实例)的细胞中毒性的实施方案。
b.表征测试化合物的基于时间的肝毒性的方法
在一些实施方案中,本发明提供了表征测试化合物的基于时间的肝毒性的方法。测试化合物可以不限于任何类型的化合物,包括具有或正在研究用于治疗目的的化合物以及环境和工业化学品。
在一些实施方案中,所述方法包括将包含肝细胞的第一体外培养物与测试化合物一起温育第一培养时间。在一些实施方案中,将包含肝细胞的多种第一体外培养物与测试化合物一起温育第一培养时间。测试化合物可以在第一培养时间的一个或多个时间点添加到第一体外培养物(即使用测试化合物给药第一体外培养物)。通常,培养物的至少一个特征在多个第一体外培养物之间是不同的。例如,不同的第一体外培养物可以在不同浓度的测试化合物的存在下和/或通过改变测试化合物给药的频率进行温育。
在一些实施方案中,所述方法包括在第一培养时间内测量测试化合物对第一体外培养物的肝细胞的至少一种细胞毒性效应(例如,在第一培养时间结束时测量),由此确定测试化合物在第一次培养时间内的肝脏毒性。如果该方法在不同浓度的测试化合物下使用多种包含与测试化合物温育的肝细胞的第一体外培养物达第一培养时间,则该方法通常包括测量测试化合物在第一培养时间内对多种第一体外培养物中的每一种的肝细胞的至少一种细胞毒性效应。在一些实施方案中,随后在第一次培养时间内确定测试化合物的tc50值。
在一些实施方案中,所述方法还包括将包含肝细胞的第二体外培养物与测试化合物温育第二培养时间。第二培养时间通常为比第一培养时间更长的持续时间。在一些实施方案中,第二培养时间比第一培养时间包含更大数量的测试化合物的个体剂量。在一些实施方案中,将包含肝细胞的多种第二体外培养物与测试化合物温育第二培养时间。测试化合物可以在第二次培养时间的一个或多个时间点添加到第二次体外培养物中。通常,培养物的至少一个特征在多种第二体外培养物间不同。例如,不同的第二种体外培养物可以在不同浓度的测试化合物的存在下和/或通过改变测试化合物给药的频率来温育。
在一些实施方案中,所述方法包括测量测试化合物在第二次培养时间(例如在第二次培养时间结束时)对第二次体外培养物的肝细胞的至少一种细胞毒性效应,以由此确定测试化合物在第二次培养时间内的肝毒性。如果所述方法在不同浓度的测试化合物下利用与测试化合物温育的包含干细胞的多种第二体外培养物达第二培养时间,则所述方法通常包括测量测试化合物在第二培养时间内对多种第二体外培养物的干细胞的至少一种细胞毒性效应。在一些实施方案中,随后在第二培养时间内确定测试化合物的tc50值。在一些实施方案中,所述方法包括在至少一个另外的培养时间结束时测量测试化合物对至少一种额外的体外培养物的肝细胞的至少一种细胞毒性效应(即持续时间甚至长于第二培养时间的持续时间的培养时间),以由此确定测试化合物在第三培养时间内的肝毒性。
通常,测试化合物在第一和第二培养时间内的肝毒性定义为在第一和第二培养时间完成时确定的测试化合物的tc50值。在一些实施方案中,通过对活细胞和/或死细胞进行计数的方法来确定存活和/或死亡的细胞的数量。在一些实施方案中,使用测量至少一种毒性过程的发生程度和/或速率的方法。在一些实施方案中,在每个这样的培养时间结束时测量的测试化合物的肝毒性被定义为测试化合物在施用特定剂量至人或其他哺乳动物的血浆中达到的最大浓度(称为“cmax”浓度)除以tc50值的比率,表示为tc50/cmax。
将测试化合物在第一次培养时间内的肝毒性与测试化合物在第二次培养时间内的肝毒性进行比较,以由此表征测试化合物基于时间的肝毒性。在一些实施方案中,该比较是测试化合物在第一培养时间内的毒性程度与测试化合物在第二培养时间内的毒性程度的简单算术比率。在一些实施方案中,测试化合物在第二次培养时间内的肝毒性大于测试化合物在第一次培养时间内的肝毒性,并且测试化合物被鉴定为展示基于时间的肝毒性,这样在第一(即,较短持续时间)培养时间结束时测量的tc50值除以在第二(即,较长持续时间)培养时间结束时测量的tc50值的算术计算是大于一(1)的数字。本发明部分基于发明人令人惊讶的观点,即相比简单测量在第二(即,较长持续时间)培养期结束时测量的毒性,该比较是测试化合物体内毒性的更精确的预测因素。该结果是出人意料的,且与本领域的常规观点相悖。
在一些实施方案中,将在第二培养时间结束时测量的测试化合物的肝毒性与在第一培养时间结束时测量的测试化合物的肝脏毒性之间的计算比率、差异或变化与定义的阈值进行比较,以确定是否观察到确定的体内毒性风险。在一些实施方案中,阈值是预定义的阈值。在一些实施方案中,阈值是绝对值(例如至少为4的比值),而在其他实施方式中,阈值是实验确定的对照化合物的比率的倍数。
在一些实施方案中,测试化合物在第二次培养时间内的肝毒性比测试化合物在第一次培养时间内的肝毒性大至少预定义的阈值,且测试化合物被鉴定为展示基于时间的肝毒性。在一些实施方案中,测试化合物在第二次培养时间内的肝毒性未比测试化合物在第一次培养时间的肝毒性大至少预定义的阈值,且测试化合物被鉴定为不显示基于时间的肝毒性。
在一些实施方案中,测试化合物在第二次培养时间内的肝毒性比测试化合物在第一次培养时间内的肝毒性大至少2倍,至少3倍,至少4倍,至少5倍,至少6倍,至少7倍,至少8倍,至少9倍或至少10倍。该倍数可以是算术计算的比值,对数衍生数值之间相减的差或任何其它形式的计算测量。
在一些实施方案中,第一培养时间为十二小时到五天。在一些实施方案中,第一培养时间是十二至三十六小时。在一些实施方案中,第一培养时间为一天到五天、一天到三天,或三天到五天。在一些实施方案中,第一培养时间为两天到四天。在一些实施方案中,第一培养时间是一天,两天,三天,四天或五天。在一些实施方案中,第一培养时间不到五天。在一些实施方案中,第一培养时间为十到六十分钟。在一些实施方案中,第一个培养时间为六十分钟到十二个小时。在优选的实施方案中,第一个培养时间是一天或十二至三十六小时。
在一些实施方案中,第二培养时间是三天到二十八天。在一些实施方案中,第二培养时间是七天到二十八天。在一些实施方案中,第二培养时间是二十八天以上。在一些实施方案中,第三培养时间比第二培养时间长。在一些实施方案中,第二培养时间是七天到十四天。在一些实施方案中,第二培养时间十四天到二十一天。在一些实施方案中,第二培养时间是二十一天到二十八天。在一些实施方案中,第二培养时间是十四天到二十八天。在一些实施方案中,第二培养时间是一,二,三,四,五,六,七,八,九,十,十一,十二,十三,或十四天,比第一培养时间长。在一些实施方案中,第二培养时间是十到六十分钟,且比第一培养时间长。在一些实施方案中,第二培养时间是六十分钟至十二个小时,且比第一培养时间长。
第二培养时间比第一培养时间长。在一些实施方案中,第二培养时间比第一培养时间长三,五,七,九,十一或十四天,或十四天以上。
第三培养时间比第二培养时间长。在一些实施方案中,第三培养时间比第二培养时间长一,二,三,五,七,九,十一或十四天,或十四天以上。
本发明包括利用简单算术、多元代数、积分、微分、概率、随机和模拟形式的测量和/或计算模型的实施方案。
c.测量测试化合物细胞毒效应的方法
在一些实施方案中,测试化合物的细胞毒性效应是细胞死亡。
在一些实施方案中,测试化合物的细胞毒性效应是导致细胞死亡的细胞毒性过程。
在一些实施方案中,所述方法包括使用多种第一培养物和不同浓度的测试化合物以获得lc50。
在一些实施方案中,细胞毒性效应是与细胞毒性特性的细胞属性。在一些实施方案中,细胞特性选自:细胞与基质的粘附、细胞内氧化还原染料成为可检测的终产物的酶促转化、活性氧(ros)的线粒体膜电位形成、细胞内酶向培养基中的释放、蛋白分泌、细胞副产物排泄和培养物中肝细胞的耗氧率。
可以使用本领域已知的任何方法评估细胞对基质的粘附。在一些实施方案中,使用由aceabiosciences,inc制造的xcelligencertcasp系统连同本文提供的肝细胞培养物来评估细胞粘附。在合适的培养基质中,例如在由aceabiosciences,inc制造的e-plateview96平板中建立肝细胞培养物。在e-培养板中电极顶部细胞的存在将影响在电极/溶液界面的局部离子环境,导致包括电极、溶液和细胞的电系统的阻抗增加。电极上附着的细胞越多,电极阻抗的增加就越大。另外,阻抗取决于细胞与电极相互作用的质量。例如,增加的细胞粘附或扩散将导致电极阻抗的较大变化。因此,显示为细胞指数(ci)值的电极阻抗可用于监测共培养物中的细胞活力、数量、形态和粘附。氧化还原染料向可检测的最终产物的细胞内酶促转化可使用本领域已知的任何方法评估。在一些实施方案中,使用来自promega的celltiter-
活性氧(ros)的形成可以使用本领域已知的任何方法来评估。在一些实施方案中,使用来自promega的ros-glotmh2o2测定。ros-glotmh2o2测定是一种均匀、快速和灵敏的生物发光测定,可以直接测量细胞培养物中或限定的酶反应中的过氧化氢(h2o2)、活性氧(ros)水平。将衍生的萤光素底物与样品一起温育,并直接与h2o2反应以产生萤光素前体。ros-glotm检测溶液的添加将前体转化为萤光素,并提供ultra-glotm重组萤光素酶以产生与样品中存在的h2o2水平成比例的光信号。该测定可以在具有或不具有血清的多种细胞培养基中进行,从而在进行测定之前不需要从培养的细胞中除去培养基。按照不需要样品处理的简单的双试剂添加方案进行均相测定。试剂添加后不到2小时即可完成测定。ros-glotmh2o2底物与h2o2直接反应,不需要辣根过氧化物酶(hrp)作为偶联酶,从而消除了与hrp抑制相关的错误命中。
检测细胞内酶向培养基释放的测定可以基于任何合适的酶并利用本领域已知的合适的试剂。在一些实施方案中,酶是腺苷酸激酶(ak)。lonza的toxilighttm生物测定试剂盒是一个合适的例子。它是一种生物发光,非破坏性的细胞溶解测定试剂盒,旨在测量受损细胞释放的酶腺苷酸激酶(ak)。ak是一种强大的蛋白质,存在于所有真核细胞中,细胞死亡时释放到培养基中。该酶主动磷酸化adp以形成atp,然后使用生物荧光萤火虫萤光素酶反应测量所得atp。随着细胞溶解水平的增加,上清液中ak的量也增加,这导致由toxilighttm试剂发射更高的光强度。由于toxilighttm生物测定试剂盒利用了ak在细胞死亡时从细胞释放的事实,因此不需要细胞裂解(不像许多其他细胞毒性测定)。因此可以随时间重复取样上清液而不破坏细胞本身。这允许细胞死亡的动力学分析。toxilighttm生物测定试剂盒还可以通过允许原始细胞进行其他测试来促进高含量筛选。ak检测所需的所有组分都包含在单一试剂中,使得分析非常易于进行。该试剂盒具有出色的灵敏度,其中检测限为每个微孔10个细胞,动态范围超过5个数量级。扩展的信号稳定性也使得该测定适于高通量筛选应用中的批处理。toxilighttm生物测定试剂盒利用ak反应的循环特性,由此少量的ak酶可以生成高浓度的atp。这使得能够检测细胞毒性非常微妙的变化并减少假阴性。toxilighttm测定检测细胞培养上清液中存在的细胞ak,消除细胞裂解细胞的需求,并允许在同一样品上进行多重检测。该测定需要简单地添加单个试剂,其可以直接添加到细胞正在生长的孔中,或者添加到抽吸的培养上清液的小样品中。
肝细胞在培养中的耗氧速率可以使用本领域已知的任何合适的测定来评估。一个例子就是seahorsebiosciences的xf技术和压力测试试剂盒。该试剂盒可以实时测量活细胞中的能量利用,同时对微孔板中的线粒体呼吸和糖酵解进行定量。xf技术为研究底物利用、线粒体功能、能量消耗和微孔板中的细胞质量提供了一种强大而简单的方法,不需要其他方法中通常使用的大量细胞、烧瓶、电极、染料、放射性物质或细胞溶解。xf分析仪通过同时测量实时呼吸和糖酵解来测量细胞代谢表型,以及病理状态下两种途径之间的转换的独特能力使得能够将细胞的生理性状与分子实体所诱导的毒性联系起来。xfe分析仪以大约2-5分钟的时间间隔测量氧气消耗率(ocr)和细胞外酸化率(ecar)。ocr是线粒体呼吸的指标,而ecar主要是糖酵解的结果。ocr和ecar的实时测量是通过在微孔板内单层细胞上分离极小体积(少于7μl)的培养基来进行的。细胞氧消耗(呼吸)和质子排泄(糖酵解)导致在这个“瞬时微型腔室”中溶解氧和自由质子的浓度的快速,容易测量的变化,其通过位于细胞单层上方200微米的固态传感器探针每隔几秒测量。仪器连续测量浓度,直到变化率为线性,随后分别计算斜率以确定ocr和ecar。一旦完成测量,探针就会升起,使上面的较大介质与瞬时微型腔室中的介质混合,将细胞值恢复到基线。一个完整的药物递送系统允许在用户定义的时间间隔内按顺序添加多达四种药物。在xf测定开始之前,将细胞接种到合适的基质上,例如xf细胞培养微孔板的孔中。在测定过程中通常使用seahorse供应的不含碳酸氢盐的培养基;然而,可以设想的是,可替换的介质也可以用于协调每个孔的多个不同测定的进行。在测定过程中可以将多达两种或四种药物添加到测定盒中用于使用。因为xf测量是非破坏性的,所以可以随时间重复测量相同细胞群的代谢率,而最多可以将两种或四种不同的药物依次注入每个孔中。在完成xf测定后,可以在相同的平板上进行其他类型的生物测定,例如细胞活力测定。总的测定时间通常为35至90分钟。为了维持正常的细胞生理学,温度控制系统将xf分析仪的内部环境维持在37℃
另一种可用于测量培养物中肝细胞耗氧率的技术是luxcel公司提供的mitoxpressxtra。
在一些实施方案中,测定中表征的细胞特征包括与基质粘附的2、3、4,5、6或7个细胞或由其组成,细胞内氧化还原染料酶促转化成可检测的最终产物、形成活性氧(ros)、细胞内酶向培养基中的释放、蛋白质分泌、细胞副产物的排泄以及培养物中肝细胞的耗氧率。这七个细胞特征中的两个、三个、四个、五个、六个和七个的每种排列独立地为本发明的具体实施方案。
在一些实施方案中,所述测定还包括测定白蛋白分泌和/或尿素排泄。
在一些实施方案中,所述系统和方法包括在六种哺乳动物物种中的至少三种上进行的毒性测定。这六种物种可以选自人、非人灵长类动物、狗和大鼠、小鼠和豚鼠。
实施例
以下实施例用于更全面地描述使用本发明的方式。提供这些实施例是为了说明的目的,而不应该限制本发明的真实范围。
实施例1:原代肝细胞-基质细胞共培养物
冷冻保存的人肝细胞获自thermofisher(原lifetechnologies公司)。冷冻保存的非人灵长类动物(食蟹猴)肝细胞获自thermofisher(原lifetechnologies公司)。冷冻保存的狗肝细胞来自ivtbioreclamation。冷冻保存的大鼠肝细胞获自thermofisher(原lifetechnologies公司)。将冷冻保存的肝细胞从液氮中取出并解冻。解冻后,将细胞重悬于培养基中,并使用台盼蓝排除法确定细胞数量和细胞活力。将基质细胞在co2培养箱中传代,直到用于实验铺板。在铺板日,将细胞从平板上分离,洗涤并重悬于培养基中。使用台盼蓝排除法确定细胞数目和活力。
将肝细胞和基质细胞以每孔30,000个肝细胞的密度接种在胶原包被的96孔板中,或以可比较的密度接种在不同大小的孔中。肝细胞基本上分散在整个孔的表面。在接种之前基质细胞停滞生长。
本文附图所示为证明肝细胞-基质细胞共培养的数个实验的结果。(图号提供在图的右下角。)
实施例2:原代肝细胞-基质细胞共培养物的稳定和持久的功能性
图1-5所示的数据显示了实施例1所述的肝细胞-基质细胞共培养物的稳定、高功能性。人代谢活性以cyp特异性代谢物形成表征。通过咪达唑仑转化为1-oh咪达唑仑的速率测量cyp3a4活性。通过将右美沙芬转化为甲右美沙芬的速率来测量cyp2d6活性。通过甲苯磺丁脲转化为4-oh甲苯磺丁脲的速率测量cyp2c9活性。狗、大鼠和猴代谢活性通过香豆素的初级和次级代谢物的形成表征。通过7-羟基香豆素的形成速率测量初级代谢,并通过7-羟基香豆素葡萄糖醛酸和7-羟基香豆素硫酸盐的形成测量次级代谢。代谢物的浓度在培养物中监测1小时,并通过lc/ms/ms分析进行分析。总体而言,结果表明,代谢功能可以在延长的时间内维持在可接受的水平。
图1显示在本发明的大鼠、狗和灵长类肝细胞-基质细胞共培养物中i期酶功能是持久的。在所示时间点测量了7-羟基香豆素形成的速率。不同模式对于不同物种是明显的;然而,在每种情况中,i期酶都是功能持久的。自肝细胞接种日,大鼠和狗具有至少35天的i期活性且灵长类具有至少22天的i期活性。
图2显示在本发明的大鼠、狗和灵长类肝细胞-基质细胞共培养物中ii期酶功能是持久的。在所示时间点测量了7-羟基香豆素葡萄糖醛酸的形成速率。不同模式对于不同物种是明显的;然而,在每种情况中,ii期酶都是功能持久的。自肝细胞接种日,大鼠和狗具有至少35天的ii期活性且灵长类具有至少22天的ii期活性。
图3显示从三个不同批次的原代人肝细胞生成的肝细胞-基质细胞共培养物中持久的cyp3a4功能。cyp3a4的功能通过测量1-羟基咪达唑仑的形成速率分析。观察了基线中的一些批次比彼此差异,但自细胞接种日,全部批次都具有达35天的3a4活性。
图4显示从三个不同批次的原代人肝细胞生成的肝细胞-基质细胞共培养物中持久的cyp2c9功能。cyp2c9的功能通过测量4-羟基甲苯磺丁脲的形成速率分析。观察了基线中的一些批次比彼此差异,但自细胞接种日,全部批次都具有达35天的2c9活性。
图5显示使用人、狗、大鼠和灵长类肝细胞建立的肝细胞-基质细胞共培养物中的小管染色。
实施例3:高功能稳定性的延长窗口
图6-9所示的数据表明,实施例1中描述的肝细胞-基质细胞共培养物的稳定、高功能性存在于延长的时间窗口内,其允许在非常高的代谢功能水平和狭窄的酶活性波动的框架内进行两周的实验。人代谢活性通过cyp特异性代谢物形成表征。通过咪达唑仑转化为1-oh咪达唑仑的速率测量cyp3a4活性。通过将右美沙芬转化为甲右美沙芬的速率来测量cyp2d6活性。通过甲苯磺丁脲转化为4-oh甲苯磺丁脲的速率测量cyp2c9活性。狗、大鼠和猴代谢活性通过香豆素的初级和次级代谢物的形成表征。通过7-羟基香豆素的形成速率测量初级代谢,并通过7-羟基香豆素葡萄糖醛酸和7-羟基香豆素硫酸盐的形成测量次级代谢。在培养物中监测1小时代谢物的浓度并标准化至第1天的形成速率,并通过lc/ms/ms分析进行分析。总的来说,结果表明,代谢功能可以延长的时间保持在稳定性窗口中,且这使得实验者能够进行长期的实验而不会使细胞功能下降。
图6显示培养物中从第7天至第25天人肝细胞-基质细胞共培养物中肝细胞的cyp3a4活性是稳定的。到第7天,培养的灵长类肝细胞具有完成的重塑和稳定化的功能。至第25天功能保持稳定。
图7显示在人肝细胞-基质细胞共培养物中,在大多数或全部三个不同批次的人细胞中,培养的第7天至第18天间四种不同的cyp酶保持稳定。
图8显示对于i期代谢的多物种稳定性窗口。
图9显示对于i期代谢的多物种稳定性窗口。
实施例4:原代肝细胞-基质细胞共培养物与hepg2细胞的比较
将hepg2细胞接种到96孔板中并使其汇合24小时。根据实施例1建立原代人肝细胞共培养物,并在实验前适应环境7天。在实验开始时,将细胞暴露于不同浓度的化合物4天,在实验的第二天重复一次给药。在第四天运行promegaatp测定。结果如图10所示。对于氟他胺和酮康唑,与hepg2细胞毒性曲线相比,观察到人肝细胞共培养物毒性曲线的右移。这似乎反映了毒性母体化合物在人肝细胞共培养物中更大的肝清除率,这意味着它需要更高浓度的母体化合物以产生与hepg2细胞等同水平的毒性。对于曲格列酮,观察到与hepg2细胞相比,在原代人肝细胞共培养物毒性曲线中的左移。这被认为是由于在原代肝细胞共培养物体系中毒代谢产物产生更多,并且需要较低的母体浓度来产生等同水平的毒性。
实施例5:多剂量依赖性毒性的检测
图11-13中的图给出了人、狗、灵长类和大鼠中重复化合物暴露6天后产生的浓度依赖性毒性曲线。整个浓度范围内每个化合物每两天给药达两天、四天和六天的治疗时间。在第六天,使用promegacelltiterblue测定来测量线粒体代谢活性。该数据随后用于生成tc50值。数据表明,在每个物种中,毒性随着时间存在浓度依赖性变化。在环磷酰胺的情况下,这被认为是由于形成毒性代谢物而发生的。数据还显示不同的物种产生不同的毒性概况。图11-13所示的数据表明,使用实施例1中描述的肝细胞-基质细胞共培养物来分析延长时间的化合物毒性。在图11中,在整个浓度范围内每两天施用一次环磷酰胺达两天、四天和六天的治疗时间。数据清楚地表明药物的多次给药会改变毒性曲线,从而可以观察到较慢的毒性形成。这是在本领域已知的其他系统中不能完成的。除此之外,这些数据表明,本发明的系统和方法能够检测和证明毒素概括中的种间差异。
图11显示多剂量的环磷酰胺对包含人、狗、灵长类或大鼠的原代肝细胞的肝细胞-基质细胞共培养物的肝毒性效应。使用celltiter-blue(promega)测定分析了细胞活力,n=4。数据显示延长给药持续时间增加环磷酰胺的细胞毒性。
图12显示处理6天后(三次给药),多剂量曲格列酮对包含人、狗、灵长类或大鼠的原代肝细胞的肝细胞-基质细胞共培养物的肝毒性效应(计算的tc50)。在图12所示的三个图中分别单独比较了狗、猴和大鼠与人的结果。
图13总结了环磷酰胺和曲格列酮的结果。
实施例6:人,狗,猴和大鼠原代肝细胞-基质细胞共培养物的比较
图14给出了gsh和promegacelltiterblue测定的比较。在50*cmax完成测定。每种化合物在第0天开始每2天给药。在第6天,测定promegacelltiterblue以评估线粒体功能。非常低的批次间差异性是本发明的系统和方法有效性和能力的进一步证明。
实施例7:基于时间的肝毒性测定
原代肝细胞-基质细胞共培养物(见实施例1-5)的稳定,持久,高功能性使得能够在延长的时间内分析培养物中的肝毒性。分析具有已知体内肝毒性概况的一组二十种化合物以评估该系统预测体内肝毒性的能力。该组包括已知展示强体内肝毒性或中度体内肝毒性的化合物以及不被视为肝毒性的化合物(图15)。已知为肝毒性的化合物包括理解为展示出(1)复杂和多因素,(2)未知和/或特异体质,(3)活性代谢物介导的,(4)通过改变胆盐运输介导的5)和胆汁淤积的肝毒性的化合物。
图16和18给出了当将上述二十种化合物暴露于肝细胞-基质共培养物时的结果,所述肝细胞-基质共培养物分别包含来自人(图16)和来自大鼠(图18)物种的肝细胞。
“标准单培养物”中的人细胞为冷冻保存的人肝细胞,将其解冻并铺在96孔板中。使它们粘附24小时,然后暴露于化合物24小时。“标准单培养物”中的大鼠细胞是新鲜分离的细胞,其铺在96孔板中。允许细胞粘附24小时,且随后暴露于化合物24小时。所有的共同培养都是根据实施例1制备。在接种后第7天开始化合物给药,每48小时加入新鲜的化合物。
图16和图18所示的数据表明,在这一组化合物中,在单一时间点分析毒性并不是体内肝毒性的有利预测方式。例如,图16中的人体数据显示,在24小时,氯丙嗪的lc50为21,普萘洛尔的lc50为105。已知这些化合物在体内分别展示中度肝毒性和缺乏肝毒性。相反,波生坦的lc50是757,单独考虑表明波生坦是一种比氯丙嗪和普萘洛尔弱得多的肝毒素。然而,波生坦实际上已知表现出强体内肝毒性。类似地,7天和14天的数据显示,数种在体内没有肝毒性的化合物或表现出中度体内肝毒性的化合物的lc50值低于已知在体内展示出强肝毒性的数个化合物的lc50值。图18中给出的大鼠数据显示观察到的体外毒性与已知的体内毒性之间类似的强关联的缺乏。
更广泛地说,相关领域的技术人员普遍认为,在时间上单个时间点(例如在24小时)进行的毒性测量几乎没有预测用途,并且对于是否将化合物进入临床前或临床药物开发的后期阶段没有给予很大的可信度。例如,产生相当低tc50值(例如100或甚至50微摩尔)的测试化合物可能被忽视,并且该化合物在临床前或临床开发中向前推进,决定推进化合物将伴随着所有附带投入的时间和金钱,即使获得“阳性肝脏信号”,这是因为药物开发者希望或相信当药物是随后在临床前动物物种或人中进行测试时,单一时间点体外测试将证明并非是预测性的,或“翻译性的”。而且,在需要的临床前开发阶段,没有可以评估毒性的单一时间点测量的外部标准。一个这样的标准,即“治疗窗口”的大小或药物达到的最大血浆浓度比其tc50(即tc50/cmax)的关系在需要时是不可用的,因为临床前的测试是在美国食品和药物管理局(fda)等监管部门已将调查性新药状态授予测试化合物之前进行的阶段。因此不允许对人施用,并且不可能获得用于计算和估计治疗窗口的实验cmax值。相关的,对于从未故意作为药物施用的环境和工业化学品的毒理学测试,cmax值从未获得。
令人惊讶的是,在分析图16和18中的数据时,观察到在以下段落中阐述的某些情况下,24小时肝毒性与7天肝毒性的比率,或24小时肝毒性与14天肝毒性的比率(分别称为“24/7毒性比率信号”或“24/14毒性比率信号”或统称为“毒性比率信号”)是体内肝毒性的有利预测方式。图17比较了24小时肝毒性与7天肝毒性,24小时肝毒性比14天肝毒性和7天肝毒性比14天肝毒性。毒性比率信号由图16所示的原始lc50值形成,因此没有单位。较高的lc50值表示较低的肝毒性效价,较低的lc50值表示较高的肝毒性效价。因此,图17中较高的毒性比率信号表明较晚时间点的肝毒性大于较早时间点的肝毒性,而图17中较低的比例表明较晚时间点的肝毒性低于较早时间点的肝毒性。图19中给出了图18中所示大鼠数据的类似分析。
基于这些数据,选择4的比率作为鉴别有意义的升高的毒性比率信号,从而表明如果信号在4或更高时,测试化合物可能是体内肝毒性的。选择肝毒性阈值的挑战在于,对于毒性比率信号具有实验价值,它必须产生非常低的假阳性信号,因此不能设置得太低;但如果设置得太高,可能会开始错过真正的阳性结果,并产生随后被发现为假阴性的信号。因此,毒性比率信号阈值可以以并入本领域已知的作为预测的灵敏度(真阴性信号的高发生率)和预测的特异性(真阳性信号的高发生率)之间有用的折衷方式来选择。在实践中,平衡敏感性和特异性的阈值常常是凭经验确定的。
如图20a至20c所示,通过应用为4的毒性比率阈值(4或更高的值表示体内肝毒性的可行的可能性,小于或等于4的值则不会),将这20种化合物分成两组。该评分系统预测了十四种化合物中的五种具有已知的体内肝毒性的毒性,并且在其余九种中不预测毒性。正确地,评分系统不在未展示体内肝毒性的6种化合物的任一种中预测毒性。
值得注意的是,五种评分为阳性的化合物均展示出以直接损伤脂质双分子层或胆盐运输改变或反应性代谢物介导表征的毒性,并且100%如此表征的化合物评分为阳性。此外,评分不产生假阳性,其中非肝毒性化合物将被评分为阳性。换句话说,利用这组化合物,利用肝毒性阈值评分为4的毒性比率信号模型产生100%的特异性(真阴性的发生率)(即,没有假阳性)。总体而言,模型的灵敏度(真阳性的发生率),如在20个参考化合物的样本集上评估的,大致为36%。然而,就其中大概的或可能的毒性机制是反应性代谢物介导的或涉及胆盐转运的改变或对肝细胞脂质双层的直接损伤的样品子集而言,模型真阳性结果生成的发生率(灵敏度)为100%。上述机制都依赖于基于细胞的模型(在这种情况下,肝细胞和基质细胞的共培养)的高,稳定和长期的代谢能力并且与之相关。相反,九种评分为阴性的已知的肝毒性化合物表现出特异体质或不被充分了解的毒性。这个结果是新的,因为在早期,当具有肝细胞-基质细胞共培养的先天代谢能力的基于细胞的模型不存在时,没有能生化地产生能够产生本文的阳性毒性比率信号的条件的模型。而且结果是出乎意料的,因为直到实施本文公开的实验,没有经验基础来预测体外模型能够以高灵敏度和高特异性可靠地确定肝毒性的子集,所述肝毒性是反应性代谢物介导的或归因于胆盐运输的改变。
简而言之,在此显示毒性比率信号证明了极好的灵敏度(100%真阴性);并且当应用于其中反应性代谢物,胆盐转运改变或脂质双层损伤涉及肝毒性作用机制的化合物子集时,还表现出极好的特异性(100%真阳性)。这一发现适用于所有化学和/或分子实体的实质部分,这对开发用于临床前药物开发和环境和工业化学品的体外测试的有效工具构成了重大的飞跃。没有其他目前可用的基于细胞的体外方法提供对于进行/不进行决策等同地可操作的数据。因此,本文公开的方法和系统给出一种重要的新工具,以帮助鉴别用于进一步调查和/或开发化合物,并且实质上减少鉴别候选药物和药物开发的时间和成本,并且最重要的是,减少了晚期临床前和i期的损耗。此外,尽管本文介绍的数据包含了使用制药测试化合物产生的数据,但基于时间的毒性比率信号在评估环境和工业化学实体的安全风险方面同样具有很大的实用性,其同样严重缺乏代谢响应性体外工具。
实施例8:临床和临床前肝毒性的预测
实施例6中分析的化合物都是已被充分研究的,具有已知的肝毒性概况的公共领域参考化合物。为了在盲性、回溯研究中挑战系统和方法的用途,将毒性比率信号模型应用于一组十种候选药物化合物,其中一个是已知的阴性对照(即已知的非肝毒性)且九个已经在临床前发现阶段或i期临床试验中停止了继续的药物开发。在测试组中的九种化合物中,五种在临床前或在临床中因肝信号相关的安全性(即肝毒性)原因而中止,而四种因与安全无关的原因而中断。如图21所示,使用24h/7天的比例和4的阈值,由于“肝脏信号”安全原因停止的五种化合物中的二种(40%)的化合物通过毒性比率信号被正确鉴别(化合物e和i)。如图22所示,使用24h/14天的比例和4的阈值,由于“肝脏信号”安全原因停止的五种化合物中的三种(60%)通过毒性比率信号被正确鉴别(化合物e、h和i)。没有假阳性,因为在24/7信号和24/14信号的情况下,毒性比率信号没有将阴性对照化合物“b”或任何出于非安全相关原因停止的化合物鉴别为阳性。
这些数据表明,使用基于时间的肝毒性试验作为化合物的早期体外筛选的一部分可以减少在临床前药物发现的后期阶段和在i期临床药物开发中的与安全有关的损耗,由此物质上减少药物开发中的时间和投入成本,同时对环境和工业化学品的体外毒理学测试进行类似的改进。
尽管已经参考本发明的特定实施例描述了本发明,但是本领域技术人员应该理解,在不脱离本发明的真实精神和范围的情况下可以进行各种改变并且可以替换等同物。另外,可以对本发明的目的,精神和范围做出许多修改以适应特定情况,材料,物质组成,工艺,工艺步骤或步骤。所有这些修改意图在所附权利要求的范围内。
- 还没有人留言评论。精彩留言会获得点赞!