5‑氨基‑1,2,4‑二噻唑‑3‑硫酮的合成方法与流程
本发明属于有机化学合成领域,具体涉及5-氨基-1,2,4-二噻唑-3-硫酮的合成方法。
背景技术:
5-氨基-1,2,4-二噻唑-3-硫酮(以下简称为氢化黄原素),该物质cas登记号为:6846-35-1,其结构如式1所示:
氢化黄原素为黄色粉末状固体,因其对通常的环境条件,如:日光照射、氧气、水分等均非常稳定,黄色经久不褪,因此最早被作为一种黄色色素用于油漆调色、涂料调色和塑料制品调色,其他方面的应用不多。最近,氢化黄原素或由其简单改造而来的n酰化衍生物被发现在特定的反应条件下,可作为非常优异的硫原子供体,用来对化合物进行硫代或者硫酰化。如:美国专利申请us6096881a,公开了将氢化黄原素的氨基进行酰化后的产物用于对寡聚核苷酸中的磷原子进行硫酰化的用途,反应通式如式2,该专利申请指出采用n酰化后的氢化黄原素对聚核苷酸中的磷原子进行硫酰化,反应条件温和、收率高、杂质少;
又如:中国发明专利申请cn201410342778.0,公开了一种用氢化黄原素作为硫代试剂对r-(+)-四氢呋喃-2-甲酸进行硫代得到r-(+)-硫代四氢呋喃-2-甲酸的用途,并指出采用氢化黄原素作为硫代试剂,使得由r-(+)-四氢呋喃-2-甲酸制备r-(+)-硫代四氢呋喃-2-甲酸的反应避免了高毒性物质的使用,同时收率更好,反应式如式3。
由于氢化黄原素作为硫代试剂或者硫酰化试剂的用途的发现,导致相关领域的技术人员开始关注氢化黄原素的合成问题。
现有技术中,氢化黄原素均是采用硫氰酸盐作为原料,在酸性条件下发生三分子缩合环化的方法合成。如:张金钟等人在其所发表的《氢化黄原素的质量标准研究》(《中国现代应用药学》,2013年03期)中,给出了氢化黄原素的合成方法:将硫氰酸钾溶解于水中,于50℃滴加浓硫酸,得到氢化黄原素;又如:巴特勒等人在其所著文章《氢化黄原素:其结构及生成机理,实验与mndo计算的相关性》(xanthanehydride:itsstructureandmechanismofformation,correlationofexperimentandmndocalculations)(《化学研究杂志》(journalofchemicalresearch),1982年)中,指出硫氰酸盐在酸性条件下发生聚合生成氢化黄原素,并给出了其对该反应推测的机理。
这种以硫氰酸盐为原料合成氢化黄原素的方法存在诸多弊端:首先,硫氰酸盐在酸性条件下缩合环化制备氢化黄原素的机理虽然还不明确,但从氢化黄原素的分子式可知,其中含有三个硫原子,也就是说,至少需要三分子的硫氰酸盐才能得到一分子的氢化黄原素,可见这种合成方法从原子经济学来看非常的不经济,原料中大部分的原子没有进入产品,而是作为副产物废弃掉了,以硫氰酸钾作为原料为例,即使100%的摩尔收率,产品的重量收率也只有50%,何况在实际生产过程中,由于该反应过程中的其他副反应极多,使得能够达到的最好摩尔收率只有50%,即:实际能达到的最好的重量收率只有25%,其余75%的来自原料的重量均作为副产物废弃掉了;其次,采用该方法在反应过程中会产生大量的气体,经分析发现这种反应产生的气体中不仅含有氮气、二氧化碳,还含有硫化氢、二氧化硫、各种氮氧化物甚至氰化氢等有毒气体,巴特勒等人也在其对氢化黄原素生成机理的推测中指出:该环合反应会放出氰化氢,同时,原料硫氰酸盐在酸性条件下,本身也会发生分解变为氰化氢,不仅造成了该反应收率低下,更进一步增加了有毒气体氰化氢的排放,使得生产过程中不仅要采用多种措施吸收并防止有毒气体排放,增加生产成本,更存在剧毒气体硫化氢和氰化氢的泄露危险,危害人身安全。再次,该方法需要在大量强酸的存在下才能进行,反应完成后,需要对反应中使用的强酸进行处理,不仅费时费力,也带来了环保上的压力。
综上所述,现有技术中采用硫氰酸盐在酸性条件下缩合环化制备氢化黄原素的方法收率低下、工艺危险性高、经济性差、环保压力大。
技术实现要素:
本发明所要解决的技术问题是:如何高效、安全、低成本、环保地合成氢化黄原素。
本发明解决以上技术问题的方案是:采用不同于现有技术的原料和方法合成氢化黄原素。
本发明的发明人在实验过程中同时使用到s,s’-二硫代碳酸二甲酯(以下简称荒酸二甲酯)和硫脲的情况下意外发现生成了一种黄色的固体,经化学分析,该黄色固体是氢化黄原素,因此得到了一种以荒酸二甲酯和硫脲为原料合成氢化黄原素的新方法。该方法的反应式如式4所示。
本发明提供的氢化黄原素的合成方法如下:以荒酸二甲酯和硫脲为原料,在醇类溶剂中混合,在磺酸存在下反应,蒸馏除掉溶剂后加入砜类溶剂溶解残渣,随后加入2,3-二氯-5,6-二氰苯醌(以下简称ddq)进行反应,将反应液与水混合,过滤,过滤所得固体用乙醇洗涤,再次过滤,得到氢化黄原素。
其中,上述氢化黄原素的合成方法中,所述的荒酸二甲酯与硫脲的摩尔比例为3~0.8∶1,更优选的,荒酸二甲酯与硫脲的摩尔比例为1.3~1∶1。
其中,上述氢化黄原素的合成方法中,所述的醇类溶剂为甲醇、乙醇、正丙醇、异丙醇中的任意一种。
其中,上述氢化黄原素的合成方法中,所述的磺酸为4-甲基苯磺酸或者甲烷磺酸中的任意一种。
其中,上述氢化黄原素的合成方法中,所述的砜类溶剂为环丁砜、2,4-二甲基环丁砜、3-甲基环丁砜中的任意一种。
其中,上述氢化黄原素的合成方法中,所述的ddq与硫脲的摩尔比例为3~1∶1,更优选的,ddq与硫脲的摩尔比例为2~1.6∶1。
本发明的有益效果在于:本发明提供了一种合成氢化黄原素的新方法,该方法所用的起始原料和合成方法均与现有技术不同;本方法摩尔收率在70%以上,远高于现有技术50%左右的摩尔收率;本方法合成过程中不产生氰化氢、二氧化硫等有毒气体,因此工艺无危险性,不大量使用强酸,因此环境友好度比现有技术大大提高。
附图说明
图1:本发明实施例所合成的氢化黄原素的1h-nmr图谱(氘代二甲基亚砜);
图2:sigma-aldrich公司提供的氢化黄原素的1h-nmr图谱(氘代二甲基亚砜);
图3:本发明实施例所合成的氢化黄原素的13c-nmr图谱(氘代二甲基亚砜);
图4:sigma-aldrich公司提供的氢化黄原素的13c-nmr图谱(氘代二甲基亚砜);
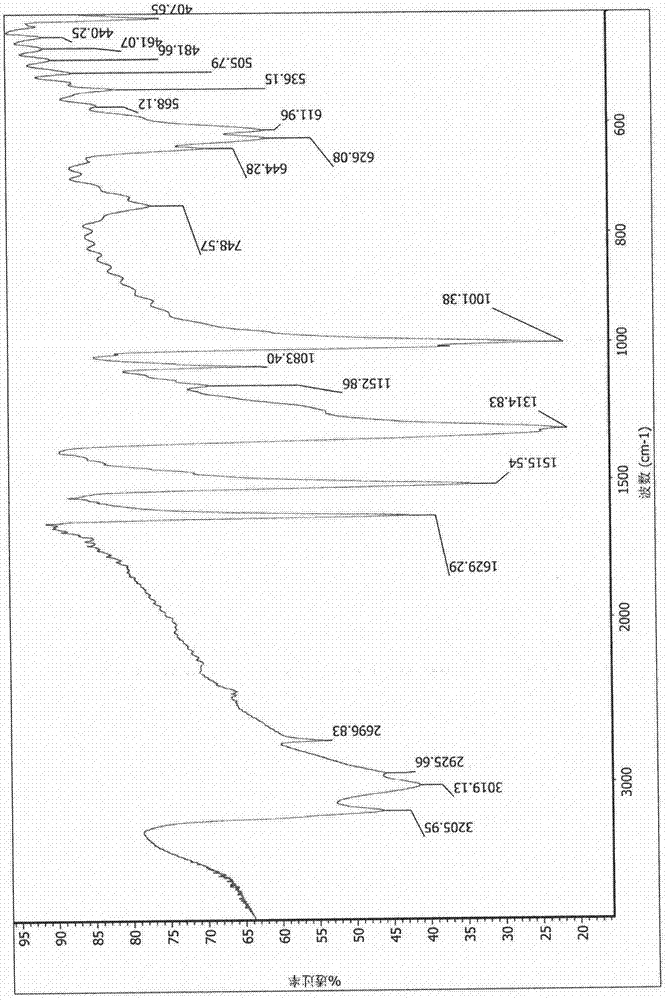
图5:本发明实施例所合成的氢化黄原素的红外光谱(溴化钾压片);
图6:sigma-aldrich公司提供的氢化黄原素的红外光谱(溴化钾压片);
具体实施方式
主要仪器与试剂:
核磁共振波谱仪:brukerdrx-400型核磁共振波谱仪;
元素分析仪:elementarvarioelcube元素分析仪;
红外光谱仪:thermoscientificnicoletis10傅立叶变换红外光谱仪;
荒酸二甲酯:sigma-aldrich公司提供;
硫脲:sigma-aldrich公司提供;
2,3-二氯-5,6-二氰苯醌(ddq):sigma-aldrich公司提供;
5-氨基-1,2,4-二噻唑-3-硫酮(氢化黄原素)对照品:sigma-aldrich公司提供。
本发明公开了一种氢化黄原素的合成方法,所述方法以荒酸二甲酯和硫脲为原料,并通过以下步骤合成氢化黄原素:
a、将荒酸二甲酯与硫脲在醇类溶剂中混合,在磺酸存在下反应;
b、蒸馏除去溶剂;
c、加入砜类溶剂溶解蒸馏残渣,随后加入ddq进行反应;
d、将反应液与水混合,随后过滤,过滤所得固体用乙醇洗涤,再次过滤,得到氢化黄原素。
其中,上述氢化黄原素的合成方法中,所述的荒酸二甲酯与硫脲的摩尔比例为3~0.8∶1,更优选的,荒酸二甲酯与硫脲的摩尔比例为1.3~1∶1。
其中,上述氢化黄原素的合成方法中,步骤a所述的醇类溶剂为甲醇、乙醇、正丙醇、异丙醇中的任意一种。
其中,上述氢化黄原素的合成方法中,所述的磺酸为4-甲基苯磺酸或者甲烷磺酸中的任意一种。
其中,上述氢化黄原素的合成方法中,步骤c所述的砜类溶剂为环丁砜、2,4-二甲基环丁砜、3-甲基环丁砜中的任意一种。
其中,上述氢化黄原素的合成方法中,所述的ddq与硫脲的摩尔比例为3~1∶1,更优选的,ddq与硫脲的摩尔比例为2~1.6∶1。
下面,通过实施例和附图对本发明的内容作出进一步的说明:
实施例1氢化黄原素的合成(荒酸二甲酯与硫脲摩尔比例的选择)
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将每份重量均为7.60g的11份硫脲分别与50ml无水乙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,分别投入重量为36.60g、30.50g、24.40g、18.30g、17.08g、15.86g、14.64g、13.42g、12.20g、10.98g、9.76g的荒酸二甲酯,投入的荒酸二甲酯与硫脲的摩尔比依次为:3∶1、2.5∶1、2∶1、1.5∶1、1.4∶1、1.3∶1、1.2∶1、1.1∶1、1∶1、0.9∶1、0.8∶1。以上反应瓶均在搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq,保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应液在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素。以上11份反应所得氢化黄原素的重量依次为:14.26g、14.31g、14.28g、14.15g、14.11g、14.19g、13.84g、13.57g、13.44g、12.06g、10.72g;摩尔收率依次为:95.1%、95.4%、95.2%、94.3%、94.1%、94.6%、92.3%、90.5%、89.6%、80.4%、71.5%。由以上数据可以看出:当荒酸二甲酯与硫脲的摩尔比例在1.3~1∶1之间时,收率较高并可兼顾经济性,继续增加荒酸二甲酯的比例,收率没有明显增高,经济性下降,而当荒酸二甲酯与硫脲的摩尔比例低于1∶1时,收率明显下降,因此,优选荒酸二甲酯与硫脲的摩尔比例为1.3~1∶1。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例2氢化黄原素的合成(ddq与硫脲摩尔比例的选择)
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将每份重量均为7.60g的8份硫脲分别与50ml无水乙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,分别向反应瓶中投入68.10g、56.75g、45.40g、40.86g、36.32g、31.78g、27.24g、22.70gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素。以上8份反应所得氢化黄原素的重量依次为:14.25g、14.22g、14.14g、14.21g、14.17g、13.09g、12.12g、10.52g,投入的ddq与硫脲的摩尔比依次为:3∶1、2.5∶1、2∶1、1.8∶1、1.6∶1、1.4∶1、1.2∶1、1∶1,摩尔收率依次为:95.0%、94.8%、94.3%、94.7%、94.5%、87.3%、80.8%、70.1%。由以上数据可以看出:当ddq与硫脲的摩尔比例在2~1.6∶1之间时,收率较高并可兼顾经济性,继续增加ddq的比例,收率没有明显增高,经济性下降,而当ddq与硫脲的摩尔比例低于1.6∶1时,收率明显下降,因此,优选荒酸二甲酯与硫脲的摩尔比例为2~1.6∶1。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例3氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水甲醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应液在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素14.13g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例4氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水乙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃续反应2小时。将反应液在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素14.20g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例5氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水正丙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应液在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素13.42g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例6氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水异丙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素13.95g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例7氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水乙醇、0.48g甲烷磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应液在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素11.29g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例8氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水乙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml2,4-二甲基环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素12.08g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例9氢化黄原素的合成
150ml三口圆底反应瓶,安装磁力搅拌和回流冷凝器,将7.60g硫脲与50ml无水乙醇、0.86g4-甲基苯磺酸投入三口圆底反应瓶中,随后,投入重量为15.86g的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100ml3-甲基环丁砜,搅拌溶解后,向反应瓶中投入40.86gddq保持30℃搅拌反应12小时,升温到50℃继续反应2小时。将反应在搅拌下倾入到200g碎冰与200ml冰水的混合物中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200ml冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素11.87g。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
实施例10氢化黄原素的合成
200l反应釜,配备回流冷凝器,将7.6kg硫脲与50l无水乙醇、0.86kg4-甲基苯磺酸投入反应釜中,随后,投入重量为15.86kg的荒酸二甲酯,搅拌下加热到回流,并保持回流反应6小时。减压蒸馏除掉反应溶剂,向蒸馏残渣中加入100l环丁砜,搅拌溶解后,向反应釜中投入40.86kgddq保持30℃搅拌反应12小时,升温到50℃续反应2小时。将反应液在搅拌下抽到装有200kg碎冰与200l冰水的混合物的500l反应釜中,继续搅拌1小时,随后过滤,将过滤所得的褐色含油状物的固体投入到200l冷却至0℃的无水乙醇中,保持0℃搅拌30分钟,再次过滤,黄色滤饼用0℃以下的无水乙醇洗涤,干燥,即得到外观为黄色结晶性粉末的氢化黄原素14.16kg。样品分析数据:1h-nmr(氘代二甲基亚砜):9.5~9.6(1h,s);9.8~9.9(1h,s);13c-nmr(氘代二甲基亚砜):183~184;208~209;红外光谱与sigma-aldrich公司提供的氢化黄原素对照品的红外光谱一致;元素分析:c15.8~15.9%,h1.3~1.4%,s64.1~64.2%,n18.6~18.7%。
- 还没有人留言评论。精彩留言会获得点赞!
- 氨基‑取代的异噻唑的制造方法与工艺
- 一种吡啶联噻唑类羧酸衍生物的合成方法与制造工艺
- 5-胡椒基-4-烷基-2-苄亚氨基噻唑及其制备方法与应用
- 5-苄基-4-烷基-2-芳氨基噻唑及其制备与应用的制作方法
- 4-叔丁基-5-(1-芳基-2-硝乙基)-2-酰氨基噻唑及其制备方法与应用的制作方法
- 4-叔丁基-5-(2-硝乙基)-2-氨基噻唑及其制备方法与应用的制作方法
- 2-芳基吗啉及其盐作为杀虫剂的应用的制作方法
- 5-[2-(苄亚氨基)噻唑-4-基]呋喃酚醚作为制备杀菌剂的应用的制作方法
- 具有杀虫活性的4-叔丁基-5-苄基-2-酰氨基噻唑及制备方法
- 具有杀虫活性的4-叔丁基-5-(1-芳基-2-硝乙基)-2-酰氨基噻唑及制备方法