一种具有光反应活性的配位聚合物及其制备方法与流程
本发明涉及一种功能性配位聚合物及其制备方法,具体的说是一种具有光反应活性的配位聚合物及其制备方法。
背景技术:
光催化烯烃的固相环加成反应具有高度的区位选择性和立体选择性,利用这种方法可以合成普通条件下很难合成甚至无法合成的环丁烷衍生物。根据schmidt烯烃环加成理论,烯烃分子中烯键的距离需要在
技术实现要素:
本发明的目的是提供一种具有光反应活性的配位聚合物及其制备方法,就是利用配位聚合物作为载体,使本来不具备光反应活性的烯烃化合物中的烯键距离拉近并且有序地平行排列,在紫外光催化下发生环加成反应,而太阳光不能催化该配位聚合物中烯烃的环加成反应。进一步利用溶剂插层,促使配位聚合物结构重组后烯键的距离更近,使其光反应活性增强,在太阳光催化下即可获得产物。所要解决的技术问题是有效地选择光敏配位聚合物的合成方法、合成途径及能产生溶剂插层的有效溶剂。
本发明所述烯烃化合物是指1,2-双(3-吡啶基)-乙烯,其化学结构如下:
1,2-双(3-吡啶基)-乙烯的晶体堆积结构见图1。相邻烯烃分子的双键间距离为
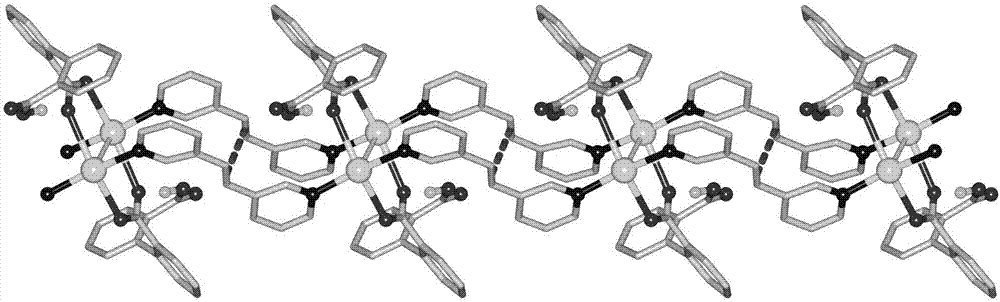
本发明具有光反应活性的配位聚合物,为配位聚合物1——[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]n或配位聚合物3——{[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]·乙腈·水}n;配位聚合物1发生光化学反应形成配位聚合物2——[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]n,配位聚合物3发生光化学反应形成配位聚合物4——{[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]·乙腈·水}n。
其中n为重复单元数,是任意正整数,表示结构单元多次重复延伸。
本发明具有光反应活性的配位聚合物的制备方法,包括如下步骤:
步骤1:以乙醇为溶剂,将1,2-双(3-吡啶基)-乙烯、2,2’-联苯二甲酸和醋酸银混合并于室温下搅拌反应8小时;反应结束后过滤,将滤液于室温下挥发除去溶剂后(约3天)可得配位聚合物1——[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]n的晶体;
步骤1中,1,2-双(3-吡啶基)-乙烯,2,2’-联苯二甲酸和醋酸银的摩尔比为1:1:1。
步骤2:将配位聚合物1置于发射波长为365nm的led灯(30w)下2cm处光照24小时,配位聚合物1的晶体碎裂,该现象说明晶体内部发生了由化学反应导致的剧烈原子振动,通过核磁共振氢谱发现配位聚合物1中的1,2-双(3-吡啶基)-乙烯完全转化为1,2,3,4-四(3-吡啶基)环丁烷,通过紫外光催化完全生成配位聚合物2——[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]n;本步骤是固相反应,没有溶剂参与,反应的收率是100%。
步骤3:将配位聚合物1置于乙腈和水的混合溶液中浸泡48小时后取出,即可得到配位聚合物3——{[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]·乙腈·水}n的晶体;本步骤的反应收率是100%。所述乙腈和水的混合溶液中乙腈和水的体积比为1:1。
步骤4:将配位聚合物3置于太阳光下光照2小时,即可得配位聚合物4——{[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]·乙腈·水}n的晶体。本步骤是固相反应,没有溶剂参与,反应的收率是100%。太阳光照强度≥70000lux,2小时即可完成反应。
纯晶态1,2-双(3-吡啶基)-乙烯没有光反应活性,本发明利用配位聚合物为载体,经紫外光催化,可使1,2-双(3-吡啶基)-乙烯发生二聚环加成反应,而太阳光不能使该反应发生;本发明进一步利用溶剂插层,促使配位聚合物中1,2-双(3-吡啶基)-乙烯的光反应活性增强,可再生、无污染的太阳光即可使1,2-双(3-吡啶基)-乙烯的二聚环加成反应发生。整个反应工艺操作简单、收率高、反应条件温和。该方法适用于高效地合成环丁烷衍生物。
本发明配位聚合物1中的金属-羧酸单元可以将1,2-双(3-吡啶基)-乙烯分子有序排列,相邻烯烃双键间的距离为
本发明配位聚合物3中,由于溶剂分子乙腈和水进入晶体的晶胞内,使得配位聚合物的结构重排,双键间的距离拉近至
配位聚合物3的光反应活性相对于配位聚合物1有了明显提升。
本发明首次利用溶剂插层显著提高烯烃化合物的光反应活性。整个反应工艺操作简单、收率高、反应条件温和。该方法适用于高效、环保地合成环丁烷衍生物。
本制备方法工艺简单、反应条件温和、产率高。
附图说明
图1为1,2-双(3-吡啶基)-乙烯的晶体结构图。
图2为[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]n的晶体结构图。
图3为[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]n的核磁共振氢谱图。
图4为[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]n的核磁共振氢谱图。
图5为[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]n在太阳光照射24小时后的核磁共振氢谱图。
图6为{[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]·乙腈·水}n的晶体结构图。
图7为{[银(1,2-双(3-吡啶基)-乙烯)(2,2’-联苯二甲酸氢基)]·乙腈·水}n的核磁共振氢谱图。
图8为{[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]·乙腈·水}n的晶体结构图。
图9为{[银(1,2,3,4-四(3-吡啶基)环丁烷)0.5(2,2’-联苯二甲酸氢基)]·乙腈·水}n的核磁共振氢谱图。
具体实施方式
本发明非限定实施例叙述为下:
1,2-双(3-吡啶基)-乙烯根据gordilloa等人发表的文章mechanisticstudiesonthepd-catalyzedvinylationofarylhalideswithvinylalkoxysilanesinwater:theeffectofthesolventandnaohpromoter方法合成(参见:gordilloa,
1、1,2-双(3-吡啶基)-乙烯单晶的制备
将1,2-双(3-吡啶基)-乙烯3.64g(20mmol)置于250ml反应瓶中,向其中加入乙醇100ml,不断搅拌直至全部溶解;将溶液置于室温下缓慢挥发,3天后可得无色晶体,收集晶体,在室温下晾干,得到1,2-双(3-吡啶基)-乙烯的单晶3.46g,产率95%。
1,2-双(3-吡啶基)-乙烯的晶体学参数:c12h10n2,mr=182.22,monoclinic,spacegroupp21/n,
1,2-双(3-吡啶基)-乙烯的x-射线单晶结构图见图1。
2、配位聚合物1的合成
将1,2-双(3-吡啶基)-乙烯1.82g(10mmol),2,2’-联苯二甲酸2.42g(10mmol),醋酸银1.67g(10mmol)和乙醇(120ml)加入到250ml圆底烧瓶中,反应物在室温下搅拌8小时后将溶液过滤,并收集滤液,将滤液置于室温下挥发3天后得到无色晶体,收集晶体,室温下晾干,可得配位聚合物1的晶体4.62g,产率87%。其晶体学参数如下:
配位聚合物1的晶体学参数:c26h19n2o4ag,mr=531.31,monoclinic,spacegroupp21/c,
配位聚合物1的x-射线单晶结构图见图2。
元素分析(c26h19n2o4ag):理论值(%):c,58.78;h,3.60;n,5.27;实验值(%):c,58.62;h,3.41;n,5.43。
1hnmr(400mhz,dmso-d6,298k,tms):δ=8.79(d,j=1.6hz,2h),8.48(dd,j=4.4,1.6hz,2h),8.05(m,2h),7.86(dd,j=8.0,1.2hz,2h),7.53(m,2h),7.42(m,6h),7.14(dd,j=7.6,0.8hz,2h)。见图3。
3、由配位聚合物1制备配位聚合物2
将配位聚合物1(1.06g,2mmol)置于直径为8cm的培养皿内,然后放置于发射波长为365nm的led灯(30w)下2cm距离处光照24小时,即可得到配位聚合物2的粉末1.06g,产率100%。
元素分析(c26h19n2o4ag):理论值(%):c,58.78;h,3.60;n,5.27;实验值(%):c,58.89;h,3.50;n,5.13。
1hnmr(400mhz,dmso-d6,298k,tms):δ=8.45(d,j=1.6hz,2h),8.26(m,2h),7.86(dd,j=7.6,1.2hz,2h),7.65(m,2h),7.53(m,2h),7.42(m,2h),7.17(m,4h),4.71(s,2h)。见图4。
将配位聚合物1(1.06g,2mmol)置于直径为8cm的培养皿内,然后置于太阳光下光照24小时,核磁共振氢谱显示配位聚合物1未发生反应。见图5。
4、由配位聚合物1制备配位聚合物3
将配位聚合物1(1.06g,2mmol)置于20ml试剂瓶中,然后向其中加入乙腈/水(v:v=1:1)溶液10ml,将试剂瓶密封放置48小时后过滤,将晶体取出并在室温下晾干,即可得配位聚合物3的晶体1.18g,产率100%。
配位聚合物3的晶体学参数:c28h24n3o5ag,mr=590.37,triclinic,spacegroup
配位聚合物3的x-射线单晶结构图见图6。
元素分析(c28h24n3o5ag):理论值(%):c,56.96;h,4.10;n,7.12;实验值(%):c,57.05;h,4.21;n,7.01。
1hnmr(400mhz,dmso-d6,298k,tms):δ=8.79(d,j=1.6hz,2h),8.48(m,2h),8.06(m,2h),7.86(dd,j=7.6,1.2hz,2h),7.53(m,2h),7.42(m,6h),7.14(dd,j=7.6,1.2hz,2h),2.07(s,3h)。见图7。
5、由配位聚合物3制备配位聚合物4
将配位聚合物3(1.18g,2mmol)置于直径为8cm的培养皿内,然后置于太阳光下光照2小时,即可完全转化为配位聚合物4的晶体1.18g,产率100%。
配位聚合物4的晶体学参数:c28h24n3o5ag,mr=590.37,triclinic,spacegroup
配位聚合物4的x-射线单晶结构图见图8。
元素分析(c28h24n3o5ag):理论值(%):c,56.96;h,4.10;n,7.12;实验值(%):c,56.72;h,4.02;n,7.24。
1hnmr(400mhz,dmso-d6,298k,tms):δ=8.45(d,j=2.0hz,2h),8.26(dd,j=4.8,1.6hz,2h),7.86(dd,j=8.0,1.2hz,2h),7.65(m,2h),7.53(m,2h),7.42(m,2h),7.17(m,4h),4.71(s,2h),2.06(s,3h)。见图9。
- 还没有人留言评论。精彩留言会获得点赞!