一种二芳基亚砜及砜衍生物及其制备方法和应用与流程
本发明属于药物化学领域,具体涉及一种二芳基亚砜及砜衍生物及其制备方法和应用。
背景技术:
随着人们生活节奏的加快,工作压力的提高,很多人长期处于亚健康状态,各种精神类疾病如抑郁症、焦虑症等的患病率也随之显著增加。抑郁症分为轻度、中度、重度。其中,患有重度抑郁症的病者受到的危害程度最大,临床上表现为注意力不集中、行动迟缓、情绪失落且无快感、有轻生的想法等。重度抑郁症分布在世界各地,尤其在大城市的患病率较高。重度抑郁症的病人基数大,自杀率高,且具有高百分比的遗传特质,抑郁症给患者家庭带来了巨大的经济负担及精神压力。
目前,临床针对抑郁症有以下几种治疗药物:传统三环类抗抑郁药(tricyclicantidepressants,tca)、单胺氧化酶抑制剂(monoamineoxidaseinhibitors,maoi)、以及近年来出现的一些新作用机制的药物,如选择性5-羟色胺(5-ht)再摄取抑制剂(selectiveserotoninreuptakeinhibitors,ssri)、选择性5-ht和去甲肾上腺素(ne)再摄取抑制剂(serotoninandnorepinephrinereuptakeinhibitors,snri)、ne和特异性5-ht能抗抑郁药(noradrenergicandspecificserotonergicantidepressant,nassa)等。现有临床药物对大部分抑郁症患者有效,然而,仍有30-40%的患者对现有药物产生不应答,需要辅助抗抑郁药物治疗才能缓解抑郁症状。且传统抗抑郁药物普遍起效时间长,ssris作为一线药物具有其明显的治疗效果,但许多患者对其治疗产生不应答,且一般2-4周后才出现明显的药效。
沃替西汀是抑郁症市场中一个成功的新药,由lundbeck和takeda公司联合研发并于2013年9月获美国fda批准上市,用于重度抑郁症的治疗。沃替西汀是具有新颖的多作用模式机制的抗抑郁药物。其对5-ht转运体具有选择性抑制作用,对ne和多巴胺能神经元几乎没有影响,同时对5-ht1a受体具有激动作用,对5-ht1b受体具有部分激动作用,对5-ht3a、5-ht7及5-ht1d受体具有拮抗作用,属多作用模式(multimodal)药物,具有较好的安全性、耐受性和有效性。但是,沃替西汀在体内半衰期长达66小时,起效时间长,一般服药2周后达到血药稳态浓度(worldjpharmacol,2015,4(1):17-30),并有致恶心、呕吐及性功能障碍等副作用。因此其作为一种长期使用的药物存在隐患和不足。
本发明通过对沃替西汀代谢通路的分析,设计并合成了相关氧化产物及衍生物,它们作为沃替西汀代谢产物及其类似物,具有治疗精神类疾病的有效性,并且有望在缩短起效时间、缩短体内半衰期、防止药物积蓄、提高稳定性等方面有所改善。
技术实现要素:
本发明的目的在于提供一种如式-1的二芳基亚砜及砜衍生物,具体包括1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)及1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐(式-1-2)。
本发明的另一目的在于提供一种如式-1的二芳基亚砜及砜衍生物的制备方法。
本发明的又一目的在于提供一种如式-1的二芳基亚砜及砜衍生物在制备抑郁症、焦虑症、癫痫症、精神分裂症等疾病的治疗药物中的应用。
本发明的目的通过下述技术方案实现:
本发明所述的如式-1所示的二芳基亚砜及砜衍生物的制备方法,包括如下步骤
1)将如式-2所示的沃替西汀氢溴酸盐溶于溶剂a中,加入碱a的水溶液,将ph调至9,水相用溶剂a萃取,合并有机相后用饱和食盐水洗涤,干燥,减压蒸出溶剂,得到如式-3所示的沃替西汀游离碱;
2)将如式-3所示的沃替西汀游离碱溶于溶剂b中,加入二碳酸二叔丁酯,25℃下反应1~3h,得到如式-4所示的4-[2-(2,4-二甲基苯硫基)苯基]-哌嗪-1-甲酸叔丁酯;
3)将如式-4所示的4-[2-(2,4-二甲基苯硫基)苯基]-哌嗪-1-甲酸叔丁酯在溶剂c中分别采用控制性氧化法(1)和控制性氧化法(2)进行控制性氧化;
其中,所述控制性氧化法(1)为采用1.1当量的氧化剂c,在0~5℃下进行反应,得到如式-5-1所示的4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯;所述控制性氧化法(2)为采用2.4当量的氧化剂c,在20~40℃下进行反应,得到如式-5-2所示的4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯;
4)将如式-5-1所示的4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯或如式-5-2所示的4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯溶于溶剂d中,加入氢溴酸,于50~70℃加热反应1~5h,再加入碳酸氢钠溶液调ph至2~3,室温下静置,析出白色晶体,于冰箱放置过夜,过滤,干燥,得产物如式-1-1所示的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪的氢溴酸盐或如式-1-2所示的1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪的氢溴酸盐;
制备路线如下所示:
具体地,步骤(1)中所述的溶剂a可以但不限于是乙酸乙酯、乙酸异丙酯、氯仿、二氯甲烷、甲苯等,优选溶剂a是乙酸乙酯。碱a可以但不限于是碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾等,优选碱a是碳酸氢钠。所述的沃替西汀氢溴酸盐(式-2)和碱a的摩尔比是1:1~1.2,优选摩尔比是1:1.2。
步骤(2)中所述的溶剂b可以但不限于是二氯甲烷,乙酸乙酯,氯仿等,优选溶剂b是二氯甲烷。所述的沃替西汀游离碱(式-3)和二碳酸二叔丁酯(boc酸酐)的摩尔比为1:1~1.5,优选摩尔比为1:1.2。进一步地,优选反应时间为1h。
步骤(3)中所述的溶剂c可以但不限于是二氯甲烷,乙酸乙酯,氯仿等,优选溶剂为二氯甲烷。氧化剂c可以但不限于是间氯过氧苯甲酸(mcpba),叔丁基过氧化氢(tbhp),异丙苯过氧化氢(chp),优选氧化剂c为间氯过氧苯甲酸。所述的控制性氧化法(1)中4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4)和氧化剂c的摩尔比为1:1~1.3,优选摩尔比是1:1.1。所述控制性氧化法(2)中4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4)和氧化剂c的摩尔比为1:2~2.4,优选摩尔比是1:2.4。
步骤(4)中所述的溶剂d可以但不限于是甲醇、乙醇等,优选溶剂d为乙醇。所述的4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1)或4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-2)和氢溴酸的摩尔比为1:2~10,优选摩尔比为1:4。进一步地,优选反应温度为65℃,反应时间为2.5h。
需要指出的是,本发明提供了如式-1-1所示的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及如式-1-2所示的1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐,将上述化合物用碱碱化后,可得到相应的游离碱,该游离碱可与其它有机酸或无机酸形成药学上可接受的盐,因而药学上可接受的其它酸加成盐也包含在本发明之中。
另外,本发明提供了如式-1-1所示的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及如式-1-2所示的1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐可形成不同的晶型、水合物、溶剂合物或前药。上述各种形式的衍生物也包含在本发明之中。
所述1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐作用于5-ht能神经系统,具有显著的抗抑郁活性,可用于制备抗抑郁症、焦虑症、癫痫症、精神分裂症等疾病的药物。
所述1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐可以经口服或其它方式给药。经口服给药时,采用常规的制剂技术,将该化合物与常规的药学上可接受的载体混合制成常规的固体制剂,包括颗粒剂、胶囊或片剂等;非经口服给药时,采用常规的制剂技术将其制成注射液、输液剂或栓剂等。
一种药物组合物,含有治疗有效量的上述如式-1-1所示的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及如式-1-2所示的1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐为活性成分,以及含有一种或多种药学上可接受的载体。所述药物组合物的各种剂型可以按照药学领域的常规生产方法制备,如使活性成分与一种或多种载体混合,然后将其制成所需的剂型。
所述药学上可接受的载体是指药学领域常规的药物载体,包括赋形剂、崩解剂、粘合剂、润滑剂、抗氧化剂、包衣剂、着色剂、芳香剂或表面活性剂。
所述的如式-1-1所示的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及如式-1-2所示的1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐和所述的药物组合物用于制备抗抑郁症、焦虑症、癫痫症、精神分裂症等疾病的药物。
所述的抗抑郁症、焦虑症、癫痫症、精神分裂症等疾病的药物的施用量是根据用药途径、患者的年龄、体重、所治疗的疾病和严重程度等变化来采取不同的用量。
与现有技术相比,本发明的优势在于:
(1)本发明设计合成了结构新颖的二芳基亚砜及砜衍生物,通过采用二碳酸二叔丁酯将1-[2-(2,4-二甲基苯硫基)苯基]哌嗪的游离仲胺基进行保护;再经控制性氧化反应,分别得到n-保护的亚砜及砜衍生物,后经串联的脱保护、成盐步骤,得到1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)及1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐(式-1-2)。本合成方法简单,反应时间短,操作简便。
(2)动物实验结果显示:本发明所提供的的1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐及1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐对5-ht能神经系统的抑制效果显著优于沃替西汀。
附图说明
附图1为1-[2-(2,4-二甲基苯硫基)苯基]哌嗪的核磁共振氢谱。
附图2为4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振氢谱。
附图3为4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振碳谱。
附图4为4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振氢谱。
附图5为4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振碳谱。
附图6为1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐的核磁共振氢谱。
附图7为1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐的核磁共振碳谱。
附图8为4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振氢谱。
附图9为4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯的核磁共振碳谱。
附图10为1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐的核磁共振氢谱。
附图11为1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐的核磁共振碳谱。
附图12为sd大鼠强迫游泳实验放弃挣扎时间柱形统计图。
附图13为sd大鼠悬尾实验静止时间柱形统计图。
具体实施方式
以下通过具体实施方式进一步描述本发明,但本发明不仅仅限于以下实施例。
实施例1:1-[2-(2,4-二甲基苯硫基)苯基]哌嗪(式-3)的制备
将沃替西汀氢溴酸盐(式-2,1.52g,4.02mmol)混悬于30ml乙酸乙酯中,加入饱和碳酸氢钠水溶液(20ml,4.8mmol),调ph=9,溶液变为澄清,静置、分液,水相用20ml乙酸乙酯萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相经无水硫酸钠干燥后减压蒸出溶剂得到1-[2-(2,4-甲基苯硫基)苯基]哌嗪(式-3)1.04g,收率:86.8%。1hnmr(400mhz,cdcl3)δ7.39(d,j=8.0hz,1h),7.15(s,1h),7.09–7.05(m,2h),7.03(d,j=8.0hz,1h),6.84-6.88(m,1h),6.50(d,j=8.0hz,1h),3.06(d,j=4.8hz,8h),2.36(s,3h),2.32(s,3h)见图1。
实施例2:4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4)的制备
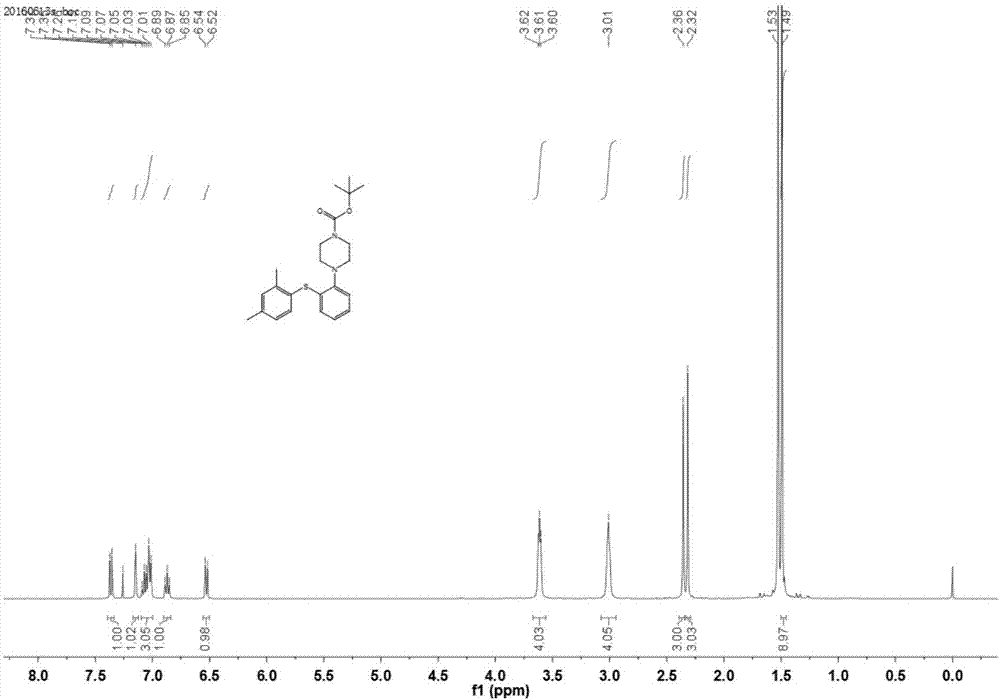
将1-[2-(2,4-二甲基苯硫基)苯基]哌嗪(式-3,1.0g,3.35mmol)溶于20ml二氯甲烷中,随后加入boc酸酐(925μl,4.02mmol),启动搅拌,25℃下反应1h。向反应瓶中加入10ml水,搅拌0.5h后静置、分液,水层以10ml二氯甲烷萃取2次,合并有机层,减压蒸出溶剂得产物1.27g,收率:95.1%。1hnmr(400mhz,cdcl3)δ7.36(d,j=8.0hz,1h),7.15(s,1h),7.10–7.09(m,3h),6.87(t,j=8.0hz,1h),6.53(d,j=8.0hz,1h),3.61(t,j=4.0hz,4h),3.01(s,4h),2.36(s,3h),2.32(s,3h),1.52(d,j=16hz,9h)见图2。13cnmr(101mhz,cdcl3)δ154.95,149.01,142.36,139.25,136.12,134.65,131.71,127.87,127.83,126.34,125.54,124.62,119.91,79.68,51.63,28.49,21.20,20.61见图3。
实施例3:4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4)的制备
将1-[2-(2,4-二甲基苯硫基)苯基]哌嗪(式-3,1.1g,3.69mmol)溶于20ml二氯甲烷中,随后加入boc酸酐(1ml,4.43mmol),搅拌,25℃下反应3h。向反应瓶中加入10ml水,搅拌0.5h后静置、分液,水层以10ml二氯甲烷萃取2次,合并有机层,减压蒸出溶剂得产物1.40g,收率:95.2%。
实施例4:4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1)的制备
将4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4,1.0g,2.51mmol)溶于20ml二氯甲烷并置于0℃的冰水浴中,将75%mcpba(0.64g,2.76mmol)溶于10ml二氯甲烷中,用恒压滴液漏斗逐滴滴加,滴加完毕后搅拌反应2h。加入饱和碳酸氢钠溶液调ph=9,静置、分液,水相用10ml二氯甲烷萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相以无水硫酸钠干燥后经柱层析(石油醚:乙酸乙酯=3:1)纯化得产物0.78g,收率:75.0%。1hnmr(400mhz,cdcl3)δ7.81(dd,j=8.0,1.6hz,1h),7.37-7.41(m,2h),7.10–7.33(m,2h),6.93(d,j=8.0hz,2h),3.36(s,2h),3.21(s,2h),2.86–2.90(m,2h),2.51(s,3h),2.32–2.34(m,2h),2.22(s,3h),1.39(s,9h)见图4。13cnmr(101mhz,cdcl3)δ154.65,150.58,141.38,140.40,136.93,131.92,131.47,127.76,127.00,126.44,126.06,122.34,79.93,52.73,28.41,21.25,19.05见图5。
实施例5:4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1)的制备
将4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4,0.5g,1.25mmol)溶于10ml二氯甲烷并置于0℃的冰水浴中,分次加入75%mcpba(0.32g,1.39mmol),反应2h。加入饱和碳酸氢钠溶液调ph=9,静置、分液,水相用8ml二氯甲烷萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相以无水硫酸钠干燥后经柱层析(石油醚:乙酸乙酯=3:1)纯化得产物325mg,收率:62.7%。
实施例6:4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1)的制备
将4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4,0.5g,1.25mmol)溶于10ml二氯甲烷并置于0℃的冰水浴中,逐滴滴加5m的tbhp癸烷溶液(276μl,1.38mmol),反应2h小时。加入饱和碳酸氢钠溶液调ph=9,静置、分液,水相用8ml二氯甲烷萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相以无水硫酸钠干燥后经柱层析(石油醚:乙酸乙酯=3:1)纯化得产物189mg,收率:36.4%。
实施例7:4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1)的制备
将4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4,0.5g,1.25mmol)溶于10ml二氯甲烷并置于0℃的冰水浴中,逐滴滴加80%chp溶液(200μl,g,1.38mmol),反应2h。加入饱和碳酸氢钠溶液调ph=9,静置、分液,水相用8ml二氯甲烷萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相以无水硫酸钠干燥后经柱层析(石油醚:乙酸乙酯=3:1)纯化得产物222mg,收率:42.7%。
实施例8:1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)的制备
将4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1,1.0g,2.41mmol)溶于10ml乙醇中,加入1.1ml48%的氢溴酸,65℃下反应2.5h,反应完成后加入10ml饱和碳酸氢钠溶液,调ph=3,室温下静置,有白色固体慢慢析出,于冰箱放置过夜,过滤、干燥后得产品832mg,收率:87.5%。1hnmr(400mhz,dmso-d6)δ8.80(s,2h),7.66(d,j=8.0hz,1h),7.59(td,j=8.0,1.6hz,1h),7.46(td,j=8.0,1.6hz,1h),7.36(d,j=8.0hz,1h),7.27(d,j=8.0hz,1h),7.17–7.14(m,2h),3.23–3.11(m,4h),2.99(s,2h),2.71-2.67(m,2h),2.44(s,3h),2.30(s,3h)见图6。13cnmr(101mhz,dmso-d6)δ151.30,141.32,141.15,140.44,136.96,131.84,131.37,127.66,126.95,126.01,125.96,122.25,54.07,46.01,21.23,19.08见图7。
实施例9:1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)的制备
将4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1,1.0g,2.41mmol)溶于6ml乙醇中,加入1.1ml48%的氢溴酸,65℃下反应1h,反应完成后加入饱和碳酸氢钠溶液,调ph=3,室温下静置,有白色固体慢慢析出,于冰箱放置过夜,过滤、干燥后得产品669mg,收率:70.3%。
实施例10:1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)的制备
将4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1,1.0g,2.41mmol)溶于6ml乙醇中,加入550μl48%的氢溴酸,65℃下反应2.5h,反应完成后加入饱和碳酸氢钠溶液,调ph=3,室温下静置,有白色固体慢慢析出,于冰箱放置过夜,过滤、干燥后得产品346mg,收率:36.4%。
实施例11:1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪盐酸盐(式-1-1)的制备
将4-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-1,1.0g,2.41mmol)溶于6ml乙醇中,加入2.0ml饱和盐酸二氧六环溶液,65℃下反应1h,反应过程中有产物白色固体慢慢析出,冷却、静置,经过滤、干燥后得产品842mg,收率:85.9%。
实施例12:4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-2)的制备
将4-[2-(2,4-二甲基苯硫基)苯基]哌嗪-1-甲酸叔丁酯(式-4,1.3g,3.26mmol)溶于20ml二氯甲烷并置于30℃水浴中,加入75%mcpba(1.93g,8.40mmol),搅拌,反应完全后以饱和碳酸氢钠溶液调ph=9,静置、分液,水相用10ml二氯甲烷萃取2次,合并有机相并以10ml饱和食盐水洗涤2次,有机相经无水硫酸钠干燥后,减压蒸出溶剂得4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯1.22g,收率:87.4%。1hnmr(400mhz,cdcl3)δ8.15(d,j=8.0,1h),8.06(d,j=8.0,1h),7.57-7.33(m,3h),7.13(d,j=8.0,1h),6.90(s,1h),3.26(s,4h),,2.70(s,4h),2.30(s,3h),2.05(s,3h),1.40(s,9h)见图8。13cnmr(101mhz,dmso-d6)δ154.81,143.65,137.77,137.49,136.37,134.71,132.84,130.89,130.32,126.31,126.02,124.49,80.01,53.12,43.08,28.44,21.34,19.65见图9。
实施例13:1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐(式-1-2)的制备
将4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-2,1.0g,2.32mmol)溶于10ml乙醇中,加入1.1ml氢溴酸,65℃下反应2.5h,反应完全后加入10ml饱和碳酸氢钠溶液,调ph=3,室温下静置冷却,有白色固体慢慢析出,于冰箱放置过夜,经过滤、干燥后得产品850mg,收率:89.2%。1hnmr(400mhz,cdcl3)δ9.18(s,2h),8.30(dd,j=8.0,1.6hz,1h),8.14(d,j=8.4hz,1h),7.67(td,j=8.0,1.6hz,1h),7.48(td,j=8.0,1.6hz,1h)7.40(dd,j=8.0,1.6hz,1h),7.33(d,j=8.0,1h),7.01(s,1h),3.11(s,8h),2.40(s,3h),2.14(s,3h)见图10。13cnmr(101mhz,dmso-d6)δ150.59,143.59,137.05,136.93,135.57,135.55,132.88,129.95,129.38,126.64,126.57,124.97,49.25,42.30,20.84,18.83见图11。
实施例14:1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪盐酸盐的制备(式-1-2)
将4-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪-1-甲酸叔丁酯(式-5-2,1.0g,2.42mmol)溶于10ml乙醇中,加入2.0ml饱和盐酸二氧六环溶液,65℃下反应1h,反应过程中有产物白色固体慢慢析出,冷却、静置,经过滤、干燥后得产品822mg,收率:86.2%。
测试例一:大鼠强迫游泳实验
实验动物:60只雄性sd大鼠,购于上海斯莱克实验动物有限责任公司,动物合格证号:(scxk)2015000535529,动物饲养于标准屏障系统动物房,适应饲养环境至少3天。
禁食情况:实验前禁食过夜,给药后4小时喂食,自由饮水。
动物分组:将大鼠分为8组,每组三只,分别为:①阴性对照(空白)组;②阳性对照(沃替西汀)组(6mg/kg);③式-1-1低剂量组(2mg/kg);④式-1-1中剂量组(6mg/kg);⑤式-1-1高剂量组(20mg/kg);⑥式-1-2低剂量组(2mg/kg);⑦式-1-2中剂量组(6mg/kg);⑧式-1-2高剂量组(20mg/kg)。
给药途径:灌胃。
动物处理:实验结束后二氧化碳安乐死。
实验过程:将37℃温水加入强迫游泳装置,液面约为装置高度70%位置,约30cm,动物置于水中开始录制视频,时长6分钟,回看视频,记录6分钟内后4分钟动物放弃挣扎时间,单位秒。
空白组大鼠放弃挣扎时间为46.3±23s,阳性对照组放弃挣扎时间为9.83±5.98s,如式-1-1所示的化合物1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐的低剂量组放弃挣扎时间为16.5±5.50s,中剂量组放弃挣扎时间为8.00±5.29s,高剂量组放弃挣扎时间为3.33±3.08s;如式-1-2所示的化合物1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸的低剂量组放弃挣扎时间为21.7±5.32s,中剂量组放弃挣扎时间为12.7±2.34s,高剂量组放弃挣扎时间为11.5±2.07s。
测试结果见附图12。
测试例二:大鼠悬尾实验
实验动物:60只雄性sd大鼠,购于上海斯莱克实验动物有限责任公司,动物合格证号:(scxk)2015000535529,动物饲养于标准屏障系统动物房,适应饲养环境至少3天。
禁食情况:实验前禁食过夜,给药后4小时喂食,自由饮水。
动物分组:将大鼠分为8组,每组三只,分别为:①阴性对照(空白)组;②阳性对照(沃替西汀)组(6mg/kg);③式-1-1低剂量组(2mg/kg);④式-1-1中剂量组(6mg/kg);⑤式-1-1高剂量组(20mg/kg);⑥式-1-2低剂量组(2mg/kg);⑦式-1-2中剂量组(6mg/kg);⑧式-1-2高剂量组(20mg/kg)。
给药途径:灌胃。
动物处理:实验结束后二氧化碳安乐死。
实验过程:用棉布固定大鼠尾部,倒挂于悬尾装置,头离桌面约10厘米,测试录制6分钟视频,回看视频进行评分,记录实验6分钟内后4分钟动物挣扎放弃的时间,单位秒。
空白组大鼠放弃挣扎时间为178±8.11s,阳性对照组(6mg/kg)放弃挣扎时间为89.5±9.29s,化合物1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)的低剂量组放弃挣扎时间为180.8±14.9s,中剂量组放弃挣扎时间为103±13.7s,高剂量组放弃挣扎时间为62.5±18.4s;化合物1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐(式-1-2)的低剂量组放弃挣扎时间为94.8±12.3s,中剂量组放弃挣扎时间为83±6.36s,高剂量组放弃挣扎时间为42.2±8.19s。
测试结果见附图13。
测试例三:大鼠脑通透性实验
实验动物:48只雄性sd大鼠,购于上海西普尔-必凯实验动物有限公司,动物合格证号:2007001670178,动物饲养于标准屏障系统动物房,适应饲养环境至少3天。
禁食情况:实验前禁食过夜,给药后4小时喂食;自由饮水。
给药途径:po-1:20mg/kg(10ml/kg)通过口服灌胃给药(n=24)
血样收集:心脏穿侧采集血液,置于提前用15%edta-k2,预处理的ep管中,以3000g,5min离心得到血浆,置于干冰环境保存,运输,再转移至-80℃冰箱存储。
脑组织收集:二氧化碳安乐死动物,确认死亡后,用剪刀剖开颈部和颅骨表层皮肤,清除颈部肌肉和结缔组织,与枕骨大孔处,用剪刀剪开枕骨,并向上掀开颅骨,暴露脑部,取出脑后,用生理盐水冲洗表面血迹,再用滤纸吸干生理盐水,称重,置于匀浆管中,加入脑重5倍体积的水,匀浆处理,得到均匀的脑组织匀浆液,置于干冰环境保存,运输,再转移至-80℃冰箱存储。
采样时间点:给药前(pre-dose)和给药后0.25,0.5,1,2,4,8,24小时,总8个时间点,终末采集血浆和脑组织样品。
实验仪器:梅特勒电子天平mettler-toledoxp26,thermofisher-70℃超低温冰箱(美国);eppendorf5415r低温冷冻离心机(德国);ikavibraxvxr小型摇床(德国);ikavortex振荡器(德国);kq5200da超声波清洗器(昆山);移液枪(gilson);vortex-genie2(奥然科技有限公司);2ml冻存管(greinerbio-one);15ml&50ml离心管(corning);注射器(上海米沙瓦医科工业有限公司)。液质联用仪(hplc-ms/ms)由api4000qtrap三重四极杆质谱仪,shimadzulc-30ad高效液相二元泵,shimadzusil-30ac自动进样器,shimadzucto-30a柱温箱以及shimadzucbm-20a脱气机组成。采用analyst5.1数据采集和处理系统。agilent1200液相色谱仪(美国):由g1312a型四元输液泵元泵,g1322a脱气机等组成。
实验过程:健康sd大鼠48只,体重190-210克,按20mg/kg的剂量给每只大鼠进行灌胃给药,大鼠给药前和给药后0.25,0.5,1,2,4,8,24小时安乐死动物,采集得到血浆和脑组织。向血浆和脑组织样品中加入10ng/ml丁螺环酮乙腈溶液沉淀蛋白,涡旋混合3min,12000rpm离心3min,取75μl上清液,加入75μl水,涡旋混合3min,再取上清10μl上清液进行lc-ms/ms分析。实验结果显示化合物1-[2-(2,4-二甲基苯亚磺酰基)苯基]哌嗪氢溴酸盐(式-1-1)的aucbrain/aucplasma值为25.4%,化合物1-[2-(2,4-二甲基苯磺酰基)苯基]哌嗪氢溴酸盐(式-1-2)的aucbrain/aucplasma值为64.3%,表明(式-1-1)及(式-1-2)两个化合物都能很好地通过血脑屏障。同时,式-1-1在脑中及血浆中的绝对暴露量更高,而式-1-2在脑中的相对暴露量更高。
表1大鼠脑通透性实验结果
测试例四:5-ht3a受体拮抗实验
1、实验材料:
①细胞:
将来源于wuxiapptec(上海)公司的鼠源m5-ht3a重组杆状病毒转导于hek-293t细胞株,在培养箱中(37℃,5%co2)孵育24小时后开始电生理试验。
②细胞外液(浴槽液)的组成:
nacl(145mm)、kcl(4mm)、cacl2(2mm)、mgcl2(1mm)、glucose(10mm),hepes(10mm),ph为7.4;渗透压调为295mosm,上述溶液的配制均使用去离子水。
③电极液的组成:
kcl(120mm)、koh(31.25mm)、cacl2(5.374mm)、mgcl2(1.75mm)、hepes(10mm)、na2-atp(4mm),用1n氢氧化钾调节ph至7.2,渗透压调至~285mosm;过滤后-20℃后保存。
④仪器:
手动膜片钳系统(axonmulticlamp700b,dididata1440,pclamp10,ect.);低通滤波设定在4khz。
2.实验过程:
①实验前准备:
将所有待测化合物(沃替西汀,式-1-1,式-1-2)与5-ht分别溶解于dmso中,配置成储备溶液,于-20℃保存。实验前用细胞外液将上述储备溶液稀释到各测试的最终浓度,并在测试前目测各溶液中是否有沉淀析出。
②手动膜片钳记录:
采用全细胞式(whole-cellmode)记录(whole-cellmode)。在细胞贴附式状态下增加负压吸引或者给予电压脉冲刺激,使电极尖端膜片在管口内破裂,即形成全细胞记录模式。膜片微电极由玻璃毛细管用电极拉制仪分两步拉制而成,充入电极内液后电极电阻为2-5mω。取出经过24小时孵育的35mm的培养皿,置于倒置显微镜载物台上,用细胞外液灌流。建立全细胞记录模式后,保持电压钳制在-60mv,在诱发的采样模型中进行记录。所有记录均在经10μm5-ht诱导出的电流稳定后进行。胞外灌流给药从低浓度开始,每个浓度给予一条刺激记录,再给下一个浓度。
使用clampfit(10.2版,moleculardevices),excel(2003版,microsoft)和graphpadprism进行进一步数据分析和曲线拟合。数据均以均值±标准差表示。在数据处理中,判断对m5-ht3a的阻断效应时,用电流的抑制率表示不同检测浓度下各化合物的作用。
ic50数值由hill方程进行拟合所得:
y:i/icontrol(即用单独施加5-ht诱导出的电流来标准化);max:为100%;min:为0%;[drug]:测试物浓度;nh:hill斜率;ic50:测试物的最大半数抑制浓度。
表25-ht3a受体拮抗实验结果
从试验结果可以看出,这两个化合物对5-ht3a受体具有明显的抑制作用。
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!