使用核酸外切酶和链移位的核酸扩增的制作方法
许多医学病症的特征在于(在患者体内)存在具有特定核苷酸序列的核酸。核酸序列可以,例如,是存在于病原细菌、病毒或已经“侵入”患者的身体并且导致患者的疾病的其他微生物的核酸序列。在许多这样的情况下,可以通过分析来自患者的样品(例如组织、血液、尿液、痰等)以确定表征微生物的核酸序列(在样品中)的存在来诊断患者体内微生物的存在。然而,在许多情况下,样品中的表征核酸序列的量非常低并且低于可检测的限度。因此,为了检测的目的,采用扩增程序来提高表征序列(或其表征变体,例如衍生自表征rrna序列的dna序列)的量。
根据本发明的第一方面,提供了一种扩增核酸序列的方法,其包括:
(a)提供扩增混合物,所述扩增混合物包含:
(i)能够实现双链核酸分子的链的消化的核酸外切酶,所述消化从所述链的5'-末端朝向3'-末端,
(ii)链移位聚合酶,
(iii)双链核酸分子,其包含彼此杂交的第一和第二核酸链,包含待扩增的第一核酸序列并具有远离其5'-末端的第一5'-末端区的所述第一链具有抵抗通过如(i)所定义的所述核酸外切酶的消化(在该方法的条件下)的核苷酸序列,
(iv)第一核酸引物,其具有与第一核酸的第一末端区相同的核苷酸序列,并且包含相同的消化抗性区,和
(v)核苷酸,其适合于供给用于扩增所述待扩增的第一核酸序列;
和
(b)在允许消化、核酸外切酶消化和链移位聚合的条件下实现扩增反应,从而产生包含扩增量的所述第一核酸序列的产物混合物。
本发明第一方面的扩增方法基于许多特征的组合。特别地,该方法利用(a)和(b)的相互关联的组合:(a)具有远离其5′-末端的消化抗性区的第一核酸引物,和(b)包含彼此杂交的第一和第二核酸链的双链核酸,所述第一链包含待扩增的核酸序列。该组合使得第一核酸链具有,从其5'-末端延伸的,与包括消化抗性序列的第一引物相同的核苷酸序列的5′-末端区。
在该扩增方法中,核酸外切酶消化第一链的5′-末端区,但不通过该链的5′-末端区中的消化抗性区。结果,第二链的3′-末端暴露并提供用于第一引物杂交的位点,其中所述第一引物在与第二链的3′-末端杂交中移位第一链的5′-末端区的未消化部分。然后第一引物通过链移位聚合酶的作用被延伸以产生第一链的拷贝。然后新合成的第一链的5′-末端区可以(通过核酸外切酶)被消化,并且所述过程实际上自我重复。
本发明的扩增方法优选在等温条件下进行,例如在45℃至55℃的温度下进行。因此,链移位聚合酶是能够在等温条件下复制模板链的聚合酶。类似地,核酸外切酶是能够在优选的等温条件下实现消化的核酸外切酶。用于本发明的优选链移位聚合酶是那些缺乏3′核酸外切酶活性的聚合酶。链移位聚合酶可以是bst系列之一,尽管存在如下所述的其他可能性。核酸外切酶优选是识别双链核酸分子的平末端的核酸外切酶。特别优选核酸外切酶是λ-核酸外切酶,其以5'和3'方向逐步降解双链dna的一条链,其优先顺序为双链结构末端的构型,即5'-凹陷的>平的>>5'-突出端,其对磷酸化而非羟基化末端优先10倍。
在本发明的一个实施方案中,双链核酸分子的第二条链可以是“正常的”,因为它不具有消化抗性区。下面参考图1更详细地描述根据该实施例的过程。在本发明的该实施方案中,优选地所述第一链的5'-末端和第二链的3'-末端一起为所述双链核酸分子提供平末端。此外,优选地所述第一链的5'-末端具有5'-磷酸基团,并且所述核酸外切酶是,在扩增混合物中优先消化在其5'-末端具有磷酸(po4)基团的双链核酸分子的链的核酸外切酶,所述消化从所述链的所述末端朝向其3'-末端,以释放第二链的3'-末端,用于引物与其杂交。在这样的实施方案中,优选地核酸外切酶是λ-核酸外切酶。
在本发明的另一个实施方案中,该方法利用具有(如第一引物)远离其5'-末端的消化抗性区的第二核酸引物。此外,在该实施方案中,第二链具有从其5'-末端延伸的5'-末端区,其具有与第二引物相同的核苷酸序列(包括消化抗性序列)。下面参考附图的图3更详细地描述该实施方案。在该实施方案中,优选地双链核酸分子具有平末端。优选地,所述第一和第二链各自的5'-末端也具有5'-磷酸基团,并且所述核酸外切酶是,在扩增混合物中优先消化在其5'-末端具有磷酸(po4)基团的双链核酸分子的链的核酸外切酶,所述消化从所述链的该末端朝向其3′-末端。优选地,核酸外切酶是λ-核酸外切酶。
对于本发明的所有实施方案,含有待扩增序列的双链dna分子可以从含有目的序列的天然存在的核酸链合成。天然存在的链可以是,例如存在于细菌或病毒中的链。天然存在的链可以是,例如rrna链。下面参考附图的图5描述从rrna链获得用于本发明方法的双链核酸构建体的程序。备选地,天然存在的链可以是dna链。在这种情况下,待根据本发明的方法扩增的双链核酸分子可衍生自变性的基因组或质粒dna(参见例如以下关于图6的描述)。
对于本发明的所有实施方案,优选地第一引物(以及第二引物,如果利用的话)包含20至30个核苷酸。相应地,第一链的5′-末端区(和,如果利用的话,第二链的5′-末端区)具有20至30个核苷酸的长度。
第一引物(和第二引物,如果利用的话)的消化抗性区优选地沿着引物的长度大约中间提供。因此,在该引物包含20个核苷酸的情况下,消化抗性区优选从自引物的5′-末端的约8-10个核苷酸开始。在引物包含30个核苷酸的情况下,消化抗性区优选从自5′-末端的约13至15个核苷酸开始。
消化抗性区可以由至少一种抵抗核酸外切酶消化的核苷酸提供。优选地,消化抗性区包含多个(例如3至6个)经修饰核苷酸的连续序列。经修饰核苷酸可以是例如硫代磷酸酯核苷酸(即其中非桥接氧原子被硫取代的核苷酸)。
如所表明的,优选地消化抗性区(例如包含3至6个经修饰核苷酸的连续序列)沿着1′引物(如果利用,2’也同样)大约中间存在。在这种情况下,特别优选地链移位聚合酶是缺乏3′核酸外切酶活性的聚合酶。然而,我们不排除消化抗性区延伸到引物的3′-末端的可能性,在这种情况下可采用具有3′核酸外切酶活性的链移位聚合酶(例如phi29)的使用。
根据本发明方法产生的扩增序列可以,例如具有每条链50至150个碱基的长度。
本发明的方法还可包含检测扩增序列的步骤。在本发明的一个优选实施例中,通过以下步骤实现检测:
(i)在所述产物混合物中提供核酸报告子组合,所述核酸报告子组合包含(a)报告链,其具有与其结合的荧光报告子部分,该报告链能够与待检测的扩增核酸序列杂交,和(b)猝灭链,其能够与所述报告链杂交并具有猝灭剂部分,所述猝灭剂部分猝灭所述荧光报告子部分的荧光,
(ii)使所述产物混合物经历变性和之后的再杂交条件,以及
(iii)检测所述荧光报告子部分的存在。
在该检测方法的步骤2(ii)中,将产物混合物相继地经历变性以及之后的再杂交条件。在变性条件下,报告链和猝灭链是产物混合物中的单独链。在再杂交条件下,报告链能够与扩增的核酸序列杂交,而不是与猝灭链杂交。因此,再杂交步骤后的荧光证实了扩增序列的存在。通常,与报告链相比,猝灭链将以摩尔过量存在。猝灭剂链与报告链的摩尔比可以是例如(1.3-1.5):1。下面结合图7更充分地描述该检测方法的实施方案。
本发明的方法特别适用于但不限于,确认生物样品(例如组织、血液、尿液、痰等)中特定的靶核酸序列的存在。靶核酸可以是例如存在于病原细菌中的靶核酸,其中所述病原细菌存在于组织样品中并且导致患者疾病。如上所概述的,并且如下文更详细描述的,从样品中提取的rrna可用于制备包含彼此杂交的第一和第二核酸链的cdna,包含证实所述生物样品中靶核酸序列的存在的第一核酸序列并具有远离其5'-末端的第一5'-末端区的所述第一链具有抗消化的核苷酸序列。然后可以使用上文更全面描述的程序扩增cdna。
这导致本发明的第二方面,根据该方面,提供了确定生物样品中靶核酸序列的存在与否的方法,该方法包括以下步骤:
(a)在下述条件下,处理所述生物样品,以便由此产生衍生物样品,所述条件为:如果所述靶核酸存在于所述生物样品中,则在所述衍生物样品中有生成的包含彼此杂交第一和第二核酸链的双链核酸分子,包含证实所述生物样品中靶核酸序列的存在的第一核酸序列且具有远离其5'-末端的第一5'-末端区的所述第一链具有抵抗消化的核苷酸序列;
(b)制备扩增组合物,其包含:
(i)能够实现双链核酸分子的链的消化的核酸外切酶,所述消化从所述链的5'-末端朝向3'-末端,
(ii)链移位聚合酶,
(iii)衍生物样品
(iv)第一核酸引物,其具有与第一核酸的第一末端区相同的核苷酸序列,如果第一核酸存在于所述衍生物样品中的话,并且包含相同的消化抗性区,和
(v)核苷酸,其适合于供给用于扩增所述待扩增的第一核酸序列;以及
(c)分析所述产物混合物中第一核酸的存在。
如上所述的本发明第一方面的所有特征均可变通适用于本发明第二方面的方法。
衍生物样品可以,例如包含从生物样品中提取的rrna制备的cdna。
仅通过举例的方式,参考附图,将进一步描述本发明,其中:
图1示意性地说明根据本发明的扩增方法的一个实施方案,以说明其基本构思;
图2示意性地说明用于图1的方法的引物;
图3a-c说明根据本发明的扩增方法的进一步的实施方案;
图4示意性地说明用于图3所示方法的正向和反向引物。
图5说明用于根据图3描述的程序,从rrna产生用于扩增的双链核酸分子。
图6描述了用于根据图3描述的程序,从基因组或质粒dna产生用于扩增的双链dna分子。
图7说明检测核酸分子的程序的实施方案。
图8显示了按照实施例1的程序合成的双链dna分子的序列。
图9说明实施例1的结果。
图10说明实施例2的结果;和
图11说明实施例3的结果。
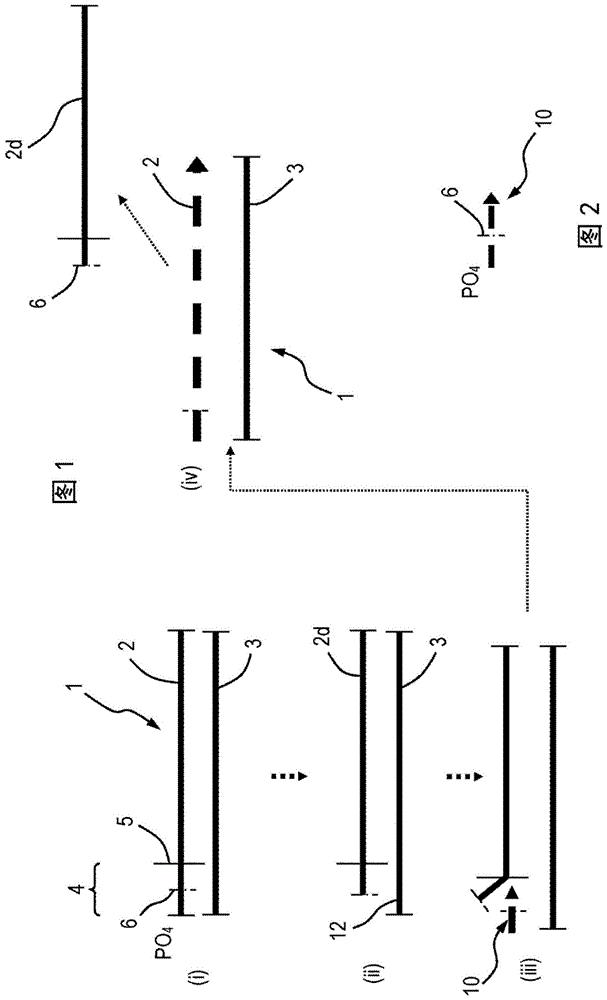
首先参考图1,其用于说明根据本发明的扩增方法当应用于其一个实施方案时的基本构思。图1中表示的是分别具有正义链2和反义链3的双链dna分子1。出于图1所示方法的目的,正义链2包含待扩增的序列。图1的方法用于该序列的线性扩增。
如图1所示,正义链2和反义链3彼此杂交并且长度相等,由此核酸分子1具有平末端。出于解释的目的,正义链2被认为具有标记为4的5′-末端区。该末端区4从正义链2的5′-末端延伸到由线5表示的点(在后面的描述中将理解其显著性的位置)。末端区的长度可以是例如10至15个核苷酸。在其5'-末端,正义链2被磷酸化,如图1中清楚描绘的磷酸基团(po4)所示。在其末端的中间,正义链2的5′-末端区4具有由线6指示的消化抗性区。该消化抗性区6可包含硫代磷酸酯(ps)核苷酸(即非桥接氧原子之一被硫原子取代的核苷酸)。虽然仅用单一线指示,但消化抗性区6通常包含若干个(例如三个)连续的经修饰的核苷酸(例如硫代磷酸酯核苷酸)。消化抗性区6可沿着末端区4大约位于中间。
现在转向反义链3,这是“平”链,因为它没有消化抗性区。另外,反义链3的5'-末端被羟基化(而不是如正义链2的情况那样被磷酸化)。
现在参考图2,其显示了用于图1的方法的引物10。引物10具有与正义链2的5'-末端区4相同的长度,并且具有与其相同的序列。因此,引物10在其5'-末端具有磷酸基团(po4)和在其末端之间相同的消化抗性区。为方便起见,引物10的消化抗性区由参考数字6表示(即标识正义链2的消化抗性区的相同附图标记)。因此,总之,引物10实际上与正义链2的5'-末端区相同。
为了进行图1的方法,制备扩增混合物,其包含双链核酸分子1、引物10、dntp、λ-核酸外切酶、链移位聚合酶(例如bst3.0)和适当的缓冲液。
在反应的最初步骤中,λ-核酸外切酶消化正义链2的5'-末端区4直至(但不超过)消化抗性区6。这是由于λ-核酸外切酶以5'-3'方向消化双链dna的一条链的“能力”,其顺序优先选择双链结构末端的构型,即5'-凹陷的>平的>>5'-突出端,其对磷酸化而非羟基化末端优先10倍。然而,λ-核酸外切酶不能通过其消化抗性区6实现正义链2的降解。为方便起见,部分消化的正义链由附图标记2d表示。该消化使得反义链3自其3'-末端在所述区域上被单链化,其中所述区域是正义链2的目前被消化部分之前与其杂交的区域。这在图1的步骤(ii)中清楚地描述-参见反义链3的左手末端,其通过附图标记12描绘了反义链3的释放的(即单链的)3'-末端区。
反义链3的释放的3'-末端区形成突出端12,其提供用于引物10杂交的靶标(参见图1的步骤(iii))。链移位聚合酶使杂交的引物10延伸以形成新的正义链2(使用反义链3作为模板),从而移位部分消化的正义链2d并再生与反义链3杂交的全长的新的正义链2(参见图1的步骤(iv))。
应当理解(由于引物10具有与图1的步骤(i)中所示的正义链2的末端区4完全相同的序列(和长度)),描述为图1步骤(iv)的结果的双链分子1与步骤(i)中所示的相同。因此,图1的步骤(iv)的产物有效地再循环到步骤(i),并且该方法连续循环以提供额外的移位链2d,由此扩增正义链2的序列。
为简单起见,图1示出了逐步机制,其中每个步骤在下一步骤开始之前“完成”。因此,例如,在从步骤(iii)到步骤(iv)的过程中,图1显示在步骤(iv)中新形成的双链核酸分子1经历通过λ-核酸外切酶的任何消化之前,完成了引物10的延伸以形成新的正义链2。然而,我们不排除在新形成的链2的消化(通过λ-核酸外切酶)开始之前引物10未完全延伸以产生完整的正义链2的可能性。
由于靶分子不再是完全双链,因此凹陷的5′-末端链将从反义链解离,其将释放出比原始λ-核酸外切酶/释放序列更长的反义序列。虽然这不能被证实,但考虑到酶靶不是完全双链的,并且本发明方法的反应可以在理论上可诱导进一步解离的相对提高的温度(例如45至55℃)下发生,因此其是可以推测的。
现在参考图3,其示出了根据本发明的扩增方法的第二实施方案。图3的方法导致指数扩增。为方便起见,图3分为图3(a)、3(b)和3(c)以便于说明本发明。首先参见图3(a),显示了双链核酸分子101,其由杂交的正义链102和反义链103组成。如在上述参考图1的双链核酸分子1的情况下,核酸分子101的正义链和反义链具有相同的长度,由此分子101具有平末端。正义链102类似于(核酸分子1的)正义链2,因为它具有5'-末端区104,其从正义链102的磷酸化5′-末端延伸到由线105描绘的点。在其末端的中间(并且大约沿其中间),5′-末端区104具有由经修饰的寡核苷酸(例如硫代磷酸酯核苷酸)形成的消化抗性区106。
核酸分子101与核酸分子1的区别在于(如图3a所示)反义链103具有从链103的5′-末端(其中存在磷酸基团(po4))延伸到线108指定位置的5'-末端区107。在其末端的中间,(反义链103的)5'-末端区107具有由线109描绘的消化抗性区。该消化抗性区可(如对于其他消化抗性区所述)包含经修饰的寡核苷酸,例如,硫代磷酸酯核苷酸。
使用如图4所示的引物110和111实现图3的反应。引物110具有对应于正义链102的5′-末端区104的序列,而引物111具有对应于反义链103的5′-末端区107的序列。这样,引物110和111二者都具有(其末端中间)消化抗性区,其分别通过附图标记106和109识别(在图4中)。
为了实现图3的反应,制备扩增混合物,其包括双链核酸分子101、引物110和111、λ-核酸外切酶、链移位聚合酶、dntp和适当的缓冲液。该反应用λ-核酸外切酶进行,从链102和103各自的5′-末端直至(但不超过)消化抗性区106和109部分消化链102和103,以产生标记为102d和103d的部分消化的链。该作用产生双链分子,其中正义链和反义链的释放的3'-末端分别形成突出端113和112。
在该方法的下一步骤中,引物110与突出端112杂交,且引物111与突出端113杂交。通过链消化聚合酶,杂交引物110和111的延伸导致标记为114和115的两个双链分子的产生。双链产物114包含正义链102的残基102d,其中所述正义链102与通过引物111的延伸生成的新链116杂交。方框6的产物包含反义链103的残基103d,其中所述反义链103与由引物110产生的新链117杂交。
双链核酸分子114的进一步处理显示在图3b中,双链分子117的进一步处理显示在图3c中。
首先参照图3b,链116通过λ-核酸外切酶从其5'-末端进行消化,以生产产物,其中链116的残基描绘为116d,链102d的3'-末端已形成引物111能够与之杂交的突出端118。
现在,引物111延伸,伴随着链116d的移位。这个阶段有两种产物。一个是双链分子114(使用链102d作为模板通过引物111的延伸产生)。现在,该双链分子在图3b的过程中有效地再循环,如箭头119所示。反应的另一产物是移位的链116d,其现在与引物110杂交。随后使用链116d作为模板延伸引物110。该延伸从左到右,如图3b所示。而且,使用引物110作为模板延伸链116d(在图3b中从右向左)。所得产物是描绘为120的双链分子,其显示为由(通过引物110的延伸形成的)杂交链121和(通过链116d的延伸形成的)122组成。
然后双链分子120的链121经历从其5′-末端直至(但不超过)消化抗性区106的消化(通过λ-核酸外切酶的作用),以提供链121d并暴露链122的3′-末端作为突出端123。如图3b所示,引物110能够与突出端123杂交并且使用链122作为模板进行延伸,伴随着链121d的移位。该步骤的产物首先是双链分子120和移位链121d。前者(即双链分子120)如箭头124所示有效地再循环,后者(即链121d)显示为与箭头125相关联,箭头125(如下所述)引起图3c所示方案的一部分。图3b中还示出了箭头126,其用于描述引入图3b的反应方案中的链116d(也在图3c中所示的方案中产生-见下文)。下面将给出对扩增过程的这个方面的进一步描述。
现在参考图3c,其实际上描述了双链分子115的处理(参见图3a),其以与图3b的反应方案相关的上文充分描述的处理双链分子114的方式处理。
因此,链117经由λ-核酸外切酶从其5'-末端进行消化,以产生部分消化的链,其与上文关于图3b描述的链121d相同。得到的双链产物包含与链103d杂交的链121d,链103d具有引物110能够其杂交的突出端130。
现在,引物110延伸,伴随着链121d的移位。这个阶段有两种产物。一个是双链分子115(使用链103d作为模板通过引物110的延伸产生)。现在,该双链分子在图3c的过程中有效地再循环,如箭头131所示。反应的另一产物是移位链121d,其现在与引物111杂交。随后使用链121d作为模板延伸引物111。该延伸从右到左,如图3c所示。而且,使用引物111作为模板延伸链121d(在图3c中从左向右)。所得产物是描绘为132的双链分子,其显示为由(通过引物111的延伸形成的)杂交链133和(通过链121d的延伸形成的)134组成。
然后双链分子132的链133经历从其5'-末端直至(但不超过)消化抗性区109的消化(通过λ-核酸外切酶的作用),以提供与图3b中(见上文)产生的链116d相同的部分消化的链,并暴露链134的3'端作为突出端135的。如图3c所示,引物111能够与突出端135杂交并且使用链134作为模板进行延伸,伴随着链116d的移位。该步骤的产物首先是双链分子132和移位链116d。前者(即双链分子132)如箭头136所示有效地再循环,后者(即链116d)显示为与箭头126相关联,箭头126(如下所述)引起图3b所示方案的一部分。图3c还示出了箭头125,其用于描述被引入图3c的反应方案中的链121d(在图3b中所示的方案中产生)。下面将给出对扩增过程的这个方面的进一步描述。
从前面的描述可以理解,图3b的程序生成单链分子121d,其也在图3c的反应方案中生成。因此,图3b显示单链分子121d如箭头125所示“传递”到图3c的反应方案。类似地,图3c导致单链分子116d的产生,其也在图3b的反应方案中产生。因此,图3c显示单链分子116d如箭头126所示“传递”到图3b的反应方案。
参考图3a-c描述的整个程序引起扩增的核酸产物(与样品中存在的原始核酸101的量相比)。
现在参考图5,其显示了用于从rrna链301产生图3(a)中所示类型的双链核酸分子101的程序的一个实施方案。如图5的方框1所示,所示反应用引物110和111实现(参见图3(a)和图4)。引物111能够与rrna链301杂交,并且引物110可以与从rrna链301产生的cdna杂交。掺入反应混合物中的用于产生cdna的是具有rna酶h活性的逆转录酶(rt)酶以及dntp和适当的缓冲液。反应在热循环条件下进行。
在该方法中,引物111与rna链301杂交,rt酶通过引物111的延伸合成互补dna(cdna)302(参见图5的方框2和3)。一旦rt酶到达rna模板链301的末端,rt酶反转方向(如箭头303所示)并且由于酶的rna酶h活性而消化rrna模板301(参见框3)。在下一步骤中,如方框4所示,引物110识别cdna序列并与其杂交。引物110通过链移位聚合酶的延伸导致框5中所示的双链构建体304,其包含与cdna链302杂交的核酸链102(参见图3(a))。双链构建体304含有目的扩增子序列加上3'cdna突出端305。现在加入用于图3中所描述的反应目的的两种酶(即λ-核酸外切酶和链移位酶诸如bst3.0)。从考虑参考图3描述的机制可以理解,cdna链302的5′-末端(远至消化抗性区109)的消化允许引物111与链102的释放的3′-末端杂交,且引物111的延伸导致双链核酸分子101的产生,伴随着cdna链302的移位(参见框6)。然后,合成的核酸构建体101以上文关于图3充分描述的方式进行扩增。
释放的cdna链不太可能在lea反应中发挥进一步的作用,因为即使引物(诸如110)会杂交并聚合,它也不会在其5′端产生平末端,因为它会凹陷(长cdna模板3′-末端突出端)。
现在参考图6,其显示了用于从(例如通过基因组或质粒dna的变性获得的)单链dna350产生用于本发明扩增程序的双链核酸分子的步骤的一个实施方案。图6的方案使用三个引物351、352和353(分别用虚线、长点划线以及中虚线描绘)。长点划线的例子是紧接在图6中的“引物”一词下面的线,并用附图标记352表示。虚线的一个例子显示在图6中“引物”一词下面的三条线的中间,并用附图标记351表示。图6中“引物”一词下面的底部的线是中虚线,并用附图标记353表示。引物351(虚线)和353(中虚线)都具有沿其各自长度的中途的消化抗性区。
两个反向引物351和352(虚线和长点划线)与链350杂交并聚合以分别产生合成链354和355(虚线和长点划线)。合成链355(长点划线)移位合成链354(虚线),以这种方式为待延伸的引物353(中虚线)提供模板,以产生合成链356,从而产生双链分子357。该双链分子357将是λ-核酸外切酶的靶标,所述λ-核酸外切酶将消化合成链354(虚线)的核酸酶敏感部分。这将释放模板链356(中虚线)的3′-末端上的序列,将允许引物351(虚线)杂交和聚合产生如图5的最后步骤中的扩增子。然后该扩增子可以进入扩增过程。同样的系统也可以用于rrna靶标以避免使用逆转录酶(rt)。例如,可以将两个反向引物351和352直接添加到rna中,并且由于bst3.0聚合酶具有逆转录作用,因此它可以产生两条链,伴随着链355(长点划线)移位链354(虚线)。因此,当链354(虚线)被移位时,它可以作为引物353(中虚线)的模板。在这种情况下,不再需要rt酶的rnaseh活性来消化rna模板以释放cdna链。
现在参考图7,其示出了用于检测根据上文关于图3描述的方法产生的双链扩增子的存在的方法的一个实施方案。
在图7中,通过图3的方法产生的扩增子由附图标记401描绘,并且显示为由杂交的链402和403(以虚线显示)组成。在扩增程序结束时,向产物混合物中加入双链核酸报告子构建体404,其由彼此杂交的核酸序列405(以点划线显示)和406(显示为实线)组成。序列405和406能够分别与扩增子401的序列402和403杂交。
如图7所示,序列405的5'-末端具有cy5分子,而序列406的3'-末端具有bhq2分子,其猝灭cy5报告子的荧光。(应当理解,可以使用其他猝灭的荧光组合)。
如图7所示,报告子构建体404具有分别在序列405和406的5'和3'-末端提供的一个平末端,并且序列405的3'-末端提供突出端。举例来说,序列405可以具有55nt的长度,而序列406可以具有35nt的长度。下面讨论这种安排的优点。
将报告子构建体404添加到扩增反应的产物中,并将混合物升高至提高的温度(例如沸点),以使双链扩增子401和双链报告子构建体404二者变性。然后使混合物冷却(例如至室温),使得序列405能够与序列402杂交,而序列406可以与序列403杂交。应当理解,在包含与序列402杂交的序列405的双链构建体中,cy5不再被猝灭,因此能够提供荧光信号以证实扩增产物的存在。应当理解,其他杂交产物是可能的(例如序列402和403可再杂交在一起,相似地序列405和406也可再杂交在一起)但是序列402和405的杂交对于检测目的是重要的。
图7中所示的检测程序的优点在于,与具有20至30nt的识别序列的分子信标(molecularbeacon)不同,由(在报告子构建体404中的)链405提供的识别序列可以是合成地尽可能长。图7中所示的程序相对于分子信标的另一个优点是分子信标的环区不具有双链体404中的竞争寡聚物(即序列406)。这也使系统更加准确。更具体地,报告序列404在热力学上更可能与正确的而非不正确的分析物序列杂交。这是因为报告序列405(具有55nt的假定长度)将优先与分析物链中相同长度的序列杂交,而不是与(较短)链405(假定为35nt)杂交。待杂交的序列405的完整55nt而不是35nt(与序列406杂交的序列405的部分)的能量需求有利于序列405与分析物序列402而不是序列406的杂交。另一方面,如果靶序列不存在于扩增混合物中,那么序列405和406之间的35nt同源性有利于这两个序列的杂交,而不是非特异性分析物和序列406之间的“更松散的键”。
本发明通过以下非限制性实施例阐释。
实施例
概要
该实施例证明了使用根据本发明的程序淋病奈瑟氏球菌(neisseriagonorrhoeae)(ng)的检测。更具体地,该实施例证明了使用根据图5的程序从ng细胞的rrna提取物产生双链dna分子,使用根据图3的程序扩增dna分子,以及使用根据图7的程序进行检测。为了说明特异性和灵敏度,dna分子的产生、扩增和检测均针对来自大肠杆菌(ec)的过量rrna的背景进行。
双链dna分子(由从ng提取的rrna产生)显示在图8中并用数字601表示。为了与图3比较,dna分子601的两条链被标记为102(seqidno5)和103。
表1显示了该实施例中使用的寡核苷酸序列。作为解释,“正向引物”序列对应于图3中的引物110,“反向引物”对应于图2中的引物111。“测定报告子”对应于图7中的序列405,且“测定猝灭剂”对应于序列406(图7中)。
表1
出于该实施例的目的,使用英国专利申请no.1609115.9中描述的程序从100×106ng细胞(通过总活菌数(tvc)确定)中提取rrna以给出200microlt洗脱液(洗脱缓冲液(eb):10mmtris-hcl,ph9.0,0.5mmedta)。使用相同的程序获得100×106ec细胞的200microlt洗脱液(通过光密度(od)测量确定)。
出于该实施例的目的,假设来自ng和ec细胞的所有rrna都收集在洗脱液中(即100%提取效率)。
程序
将源自1x106ec细胞的rna与源自250、500、1000和2000个ng细胞的rna混合,并使体积达到100microlt。详细地,从100x106ec细胞中提取的200microlt洗脱液中的2microlt将含有衍生自1x106ec细胞的rna(基于上述假设,即100%提取效率)。将2microlt的ec洗脱液的等分试样分别与ngrna的200microlt洗脱液的1/4000、1/2000、1/1000和1/500稀释的2microlt等分试样混合(该稀释液分别含有来自于250、500、1000和2000个ng细胞的rna,其提取自100×106的ng细胞起始材料,再次假设100%提取效率)。最终体积在h20中补足至100microlt,其含有:各自终浓度为50microm的两种引物(fw和rv),0.2mmdntp,4mmmgso4,20mmtris-hcl,10mm(nh4)2so4,150mmkcl,0.1%tween20,ph8.8,25℃,加0.3microlt(4.5u)的热启动逆转录酶(newenglandbiolabs,m0380)。
包括两个阴性对照情况,一个具有2microlt的eb,一个条件仅具有2microlt的ecrna(1×106ec背景对照)。每种情况一式三份。
将样品在48℃下放置10分钟,之后将5microlt酶混合物:[1microlt(8u)bst3.0dna聚合酶(newenglandbiolabs,m0374)加0.3microlt(1.5u)λ-核酸外切酶(newenglandbiolabs,m0262)加3.7microlt的h20]加入每个样品,再次将最终混合物在48℃下孵育10分钟。
在该温育结束后,将10microlt测定混合物:[报告寡核苷酸(1microlt,10pmol)加猝灭寡聚物(1.4microlt,14pmol)在含有15.2mmtris-hcl,7.6mm(nh4)2so4,114mmkcl,0.076%吐温20,ph8.8,25℃的h2o中补足至10microlt]加入各个复制子中。最终混合物在95℃温育5分钟,然后在室温下放置2分钟,转移至黑色聚碳酸酯孔中,并在荧光板读数器上对每个样品进行3次相对荧光单位(rfu)测量。
对于每个复制子,对3个读数的值进行平均,并将这些读数平均值用于统计分析。该实验独立进行9次。使用单向anova用使用未配对的学生t-检验的事后方差分析进行单向方差分析。ng特异性信号相对于非特异性106ec信号对所有ng滴定点都是显著性的(***p≤0.001)。数据点是以三份±sem进行的9个单独实验的平均值(图9.a)。图9.b显示了减去eb背景荧光对照后的所有平均值。
图9a显示了纯化的rna进入该过程后30分钟的测定结果。9个独立实验的手段的相对荧光单位(rfu)数据点以一式三份±sem进行。样品是eb(阴性背景荧光对照)、来自100万个大肠杆菌(ec)非特异性细菌的rna和掺有来自于250、500、1000和2000个淋病奈瑟氏球菌(ng)特异性细菌的rna的100万个ec。使用单向anova用使用未配对的学生t-检验的事后方差分析进行单向方差分析。ng特异性信号相对于ec非特异性信号对所有ng滴定点都是显著性的(***p≤0.001)。
图9b示出了从rfu中的所有其他样本平均值减去eb阴性背景控制平均值之后的平均值的净信号。
实施例2
该实施例提供了本发明的扩增方法与pcr的比较。
如实施例1中所述制备包含从100x106ng细胞中提取的rrna的200microlt洗脱液。
将对应于500万个ng细胞的10microlt提取物(假设100%提取效率)用2u的dnasei(newenglandbiolabs,#m0303)在1xdnasei缓冲液中(10mmtris-hcl,2.5mmmgcl2,0.5mmcacl2,ph7.6,25℃)在37℃下处理30分钟,酶在75℃下变性10分钟。这是为了确保rna提取物中不存在可能影响pcr结果的dna痕迹(pcr可以扩增16s基因以及由16srrna制备的cdna)
再次假设100%提取效率,用水稀释dnasei处理的rna提取物的等分试样,以产生代表1000000、200000、100000、50000和25000个ng细胞的样品。然后这些样品在h20中补足至100microlt的终体积,其包含:各自终浓度为50microm的两种引物(fw和rv-见表1),0.2mmdntps,4mmmgso4,20mmtris-hcl,10mm(nh4)2so4,150mmkcl,0.1%tween20,ph8.8,25℃,加0.3microlt(4.5u)的热启动逆转录酶(newenglandbiolabs,m0380)。从用于rna提取程序的1微升洗脱缓冲液(eb)以类似方式制备阴性背景荧光对照样品。
为了(a)根据本发明的扩增反应和(b)通过pcr扩增的目的,如下所述,使用1microlt等分试样的每种含有cdna的样品以及eb。
对于根据本发明的扩增程序,将1微升等分试样在h20中补足至100微升终体积,其含有:各自终浓度为1.2微摩尔的两种引物(fw和rv),0.2mmdntp,4mmmgso4,20mmtris-hcl,10mm(nh4)2so4,150mmkcl,0.1%吐温20,ph8.8,25℃,8ubst3.0dna聚合酶加1.5uλ-核酸外切酶。因此,如此制备的样品代表10000、20000、1000、500和250个ng细胞。所有样品一式两份制备(并测试)。扩增反应在48℃下进行10分钟,并将产物混合物置于冰上直至测定。
对于pcr反应,将1microlt含有cdna的样品(或eb对照)在h20中稀释至100microlt终体积,其含有:10mmtris-hcl,50mmkcl,1.5mmmgcl2,ph8.3,25℃,2.5utaq聚合酶(newenglandbiolabs,#0273),两种引物(fw和rv)各自1.2microm,和0.2mmdntps。pcr扩增方案使用13个循环实现,每个循环由在95℃的变性,在57℃的退火和在68℃的聚合组成,每个温度30秒(记录的总时间为33分钟)。将产物混合物置于冰上直至测定。
将(a)使用本发明扩增方法获得的和(b)通过pcr扩增获得的产物混合物以与上文实施例1中所述相同的方式进行测定。结果如图10(a)和(b)中所示。
图10(a)显示了使用(a)本发明方法和(b)通过pcr扩增所扩增的各种样品(和eb对照)的平均总相对荧光单位(rfu)±标准偏差(stdev)。数据点是使用两种扩增程序的测定结果的平均值(在一式两份样品上)。图10(b)显示通过从所有样品平均值中减去eb阴性对照平均值计算的净rfu值。
图10(a)和10(b)的结果显示本发明的扩增方法产生与pcr扩增方法非常相似的结果。假设100%pcr效率,13个循环的扩增比为1至8,192(213)。因此,与在33分钟pcr反应(其需要温度循环)之后获得的类似结果相比,本发明的方法产生约104的扩增比,持续10分钟的等温扩增。
实施例3
该实施例证明了缓冲液对扩增产率的影响。
使用实施例1中描述的程序,从提取物的沉淀物制备rna提取物(起始材料,使用与实施例1中所用相同的洗脱缓冲液(eb)(ph为9)洗脱的100x106个淋病奈瑟氏球菌(ng)实验室生长细胞的沉淀物)。
将2microlt稀释于eb中的等分试样洗脱稀释液(对应于总共1000个ng细胞)或2microlteb加入到81.3microlt的h20或eb或10mmtris-hclph8,0.5mmedta(“ebph8”)中,一式两份(用于补足到100microlt体积的其余体积是具有逆转录酶的h20,和浓缩形式的包括tris-hcl的lea成分,当稀释至100microlt的最终体积时,其为20mm,提供ph值8.8)。
(因此,加入81.3microlt的eb或“ebph8”时,向反应中加入了额外的8.13mmtris-hcl,使tris-hcl的最终浓度为28.13mm)。如实施例1中那样进行逆转录、扩增和测定(使用4.5u的温启动逆转录酶,8u的bst3.0dna聚合酶和1.5u的-核酸外切酶)。
结果如图11(a)和(b)所示。
在图11(a)中,数据点是一式两份扩增子用stdev的测定结果的平均值。模板是阴性对照(无rna,2microlt的ebph9)和衍生自在水、eb(ph9)或eb(ph8)中混合的1000个淋病奈瑟氏球菌(ng)实验室培养的细胞的rna。rfu代表相对荧光单位。
在图11(b)中,数据点是通过从所有样本平均值中减去eb阴性对照平均值计算的净rfu值,揭示了eb(ph8)为净rfu值提供最佳lea产率净rfu值,针对h20、eb和10mmtris-hclph8,0.5mmedta混合物分别为1626、1788和3269,因此证明了在rna提取方案中使用ebph8作为洗脱缓冲液的优点。
- 还没有人留言评论。精彩留言会获得点赞!
- 基于单链核酸外切酶末端保护分析检测小分子与结合蛋白相互作用的生物传感技术的制作方法
- 基于核酸外切酶Ⅲ/Ⅰ保护分析检测单核苷酸多态性的荧光生物传感方法
- 核酸外切酶Ⅲ消化FRET-dsDNA微阵列芯片检测转录因子蛋白的制作方法
- 人核酸外切酶因子基因编码蛋白的制作方法
- 基于t7核酸外切酶抑制分析检测有机小分子与结合蛋白相互作用的荧光生物传感方法
- 基于核酸外切酶Ⅰ保护分析检测有机小分子和蛋白质相互作用的可视传感方法
- 一种定量核酸外切酶i活性测定方法
- 一种t5核酸外切酶在大肠杆菌中的制备方法
- 限制性内切酶和核酸外切酶ⅲ的无标记荧光检测dna甲基化和甲基转移酶活性的检测方法
- 核酸外切酶保护dna探针杂交dna微阵列芯片检测dna结合蛋白的制作方法
- 基于核酸外切酶Ⅲ末端保护分析检测小分子与结合蛋白相互作用的荧光生物传感技术的制作方法
- 基于单链核酸外切酶末端保护分析检测小分子与结合蛋白相互作用的生物传感技术的制作方法
- 基于核酸外切酶Ⅲ/Ⅰ保护分析检测单核苷酸多态性的荧光生物传感方法
- 核酸外切酶Ⅲ消化FRET-dsDNA微阵列芯片检测转录因子蛋白的制作方法
- 人核酸外切酶因子基因编码蛋白的制作方法
- 基于t7核酸外切酶抑制分析检测有机小分子与结合蛋白相互作用的荧光生物传感方法
- 基于核酸外切酶Ⅰ保护分析检测有机小分子和蛋白质相互作用的可视传感方法
- 一种定量核酸外切酶i活性测定方法
- 一种t5核酸外切酶在大肠杆菌中的制备方法
- 限制性内切酶和核酸外切酶ⅲ的无标记荧光检测dna甲基化和甲基转移酶活性的检测方法