一种伊卢多啉手性中间体的化学-生物制备方法与流程
本发明涉及有机合成领域,尤其涉及一种伊卢多啉手性中间体的化学-生物制备方法。
背景技术:
2017年5月27日,美国fda批准furiex制药公司的腹泻型肠易激综合征新药伊卢多啉(eluxadoline)上市,商品名viberzi。伊卢多啉作为mu-阿片受体激动剂,作用于mu-阿片受体从而发挥治疗腹泻型肠易激综合征(ibs-d)的作用。是一种首创的口服有效、局部作用治疗药物,具有一种独特的作用机制;该药具有混合的阿片受体活性,是μ受体拮抗剂,也是delta阿片受体激动剂和kappa受体激动剂。药物进食时服用,能够显著减轻ibs-d患者的腹痛及腹泻症状。伊卢多啉分子结构如式i所示:
在伊卢多啉的合成工艺中,式ii所示的化合物(s)-2-叔丁氧羰基氨基-3-(4-氨甲酰基-2,6-二甲基苯基)丙酸为伊卢多啉api合成的关键中间体。
迄今为止,已经有多篇文献报道了该化合物的合成方案:
路线一:wo2003092688(式iii)
该路线以boc-l-2,6-二甲基络氨酸甲酯为原料经4步合成目标化合物,但起始原料结构复杂不易制备,生产成本较高。
路线二:wo2006099060(式iv)
该路线步骤2小分子与芳香环接枝收率较低,且破坏boc保护的胺基,副产物较多,分离纯化比较复杂。步骤3手性纯度低,影响最终化合物的ee值。
路线三:wo2010062590(式v)
该路线为伊卢多啉原研公司给出的制备方案,但存在收率较低,手性纯度不高的问题。
该专利还提供另一种合成路线如式vi:
该路线用到贵金属催化剂且耦合反应收率较低,不适于公斤级、吨级工业放大生产。
因此,需要针对现有技术中所存在的缺陷,对现有技术加以改进,提供一种原料易得、工艺简洁、操作方便、收率更高的制备方法,以降低成本。
技术实现要素:
本发明所要解决的技术问题是提供一种伊卢多啉手性中间体的化学-生物制备方法,解决现在(s)-2-叔丁氧羰基氨基-3-(4-氨甲酰基-2,6-二甲基苯基)丙酸生产成本高、操作复杂、产物手性纯度低的问题。
技术方案
一种伊卢多啉手性中间体的化学-生物制备方法,步骤包括:
合成路线如式vii所示:
(1)化合物3通过negishi反应得到含有中间态化合物4的锌试剂溶液,然后与化合物5发生偶联反应,得到化合物6;
(2)化合物6依次经过酰胺水解酶、氰基水合酶分别催化水解,转换为化合物7。
进一步,步骤(1)所述negishi反应步骤包括:
a.将锌粉置于无水甲醇中加热活化20~100min,然后真空干燥除去溶剂;
b.将活化后的锌粉加入无水无氧dmf,惰性气体保护下加入1,2-二溴乙烷并升温至80~90℃,然后冷却至10~30℃后加入三甲基氯硅烷;
c.分批加入化合物3的dmf溶液,控制温度10~30℃,得到锌试剂溶液;
d.配制化合物5的无水无氧dmf溶液,加入钯催化剂及配体,并升温至70~100℃,将锌试剂溶液逐步滴入,80~100℃下直至反应结束。
进一步,所述步骤(1)中,化合物3、zn粉、化合物5、钯催化剂摩尔量之比为1:1~8:0.6~1.5:0.01~0.1;所述步骤(1)中锌粉、1,2-二溴乙烷、三甲基氯硅烷的摩尔量之比为1:0.01~0.1:0.01~0.1;所述配体选自pph3、p(o-tolyl)3、dppe、dppp、dppb、dppf、binap、diop、chiraphos。
进一步,步骤(2)包括:
e.将化合物6置于酰胺水解酶缓冲溶液中水解掉分子中的酰胺;
f.高温灭活酰胺水解酶,再加入氰基水合酶,催化反应得到化合物7。
进一步,步骤(2)所述高温灭活酰胺水解酶步骤包括将反应液瞬间加热至沸腾,保持0.5~2min,然后冷却至室温。
进一步,所述化合物3制备方法包括如下反应步骤:
g.化合物1经过氨酯交换反应,得到化合物2;
h.化合物2在三苯基膦、咪唑催化下与碘发生取代反应得到化合物3。
进一步,所述步骤g具体包括:化合物1加入醇氨溶液中,80~120℃闷罐反应直至反应结束。
进一步,所述步骤h具体包括:碘分批加入溶有三苯基膦、咪唑的有机溶液,然后0~5℃下逐步滴入化合物2的溶液,搅拌至反应完成,得到化合物3。
进一步,所述步骤h中化合物2、碘、三苯基膦、咪唑摩尔量之比为1:1~2.5:1~3:1~3。
有益效果
本发明相对于现有技术,对化合物2中的羧基采用酰胺化进行保护,有效克服negishi反应中boc酰胺被破坏,导致negishi反应收率低,副产物多的问题,有效提高了反应的收率。如果以化合物3为起始原料,制备目标化合物7的整体收率不低于78.6%;如果以化合物1为起始原料计算,制备目标化合物7的整体收率不低于50.3%,相比于现有技术最高20%左右的收率来说,是明显提高的。
本发明路线化合物6制备7的过程中,采用在水相两步酶催化反应反应条件温和、绿色环保,且采用一锅法制备,后处理较为方便,整体收率在97%左右。后两步生物酶对产物的手性选择性较好,一但选择到合适的生物酶,后两步反应无需担心副反应、可逆反应等微观分子反应机理改变产物ee值的问题。
本发明路线六步反应中,仅有两步产物需要单独纯化,其余步骤均可直接一锅法或者减压干燥后直接投入下一步反应,大大节省了反应后处理成本,加快生产速度,提高生产效率。
附图说明
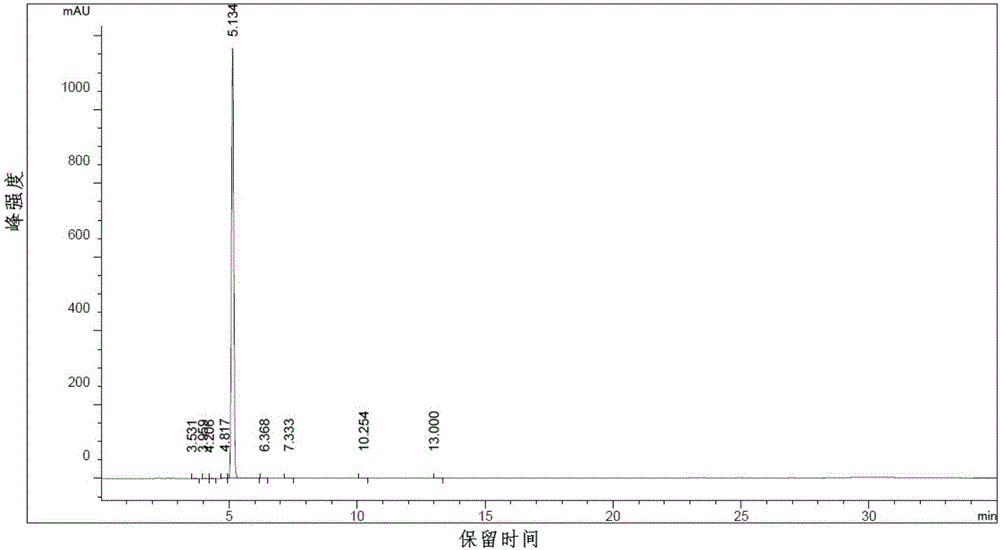
图1为本发明所制备目标化合物hplc分析图谱;
图2为本发明所制备目标化合物手性hplc分析图谱。
具体实施方式
下面结合具体实施例和附图1、2,进一步阐述本发明。
本发明是提出一种伊卢多啉手性中间体的化学-生物制备方法,具体工艺路线如式ix所示:
每一步合成工艺所对应的实施例如下:
实施例1化合物2的制备
将化合物1(22g,0.1mol)溶于甲醇220g,将氨气通入上述混合液直至饱和,置于聚四氟乙烯内衬的闷罐加热到80℃反应24h,检测反应进行完全后停止加热,冷却至室温,将反应液减压除去溶剂,所得产物直接投下一步,无需进一步纯化。
实施例2化合物3的制备
将三苯基磷(26.2g,0.1mol),咪唑(6.8g,0.1mol)溶于340ml二氯甲烷,将碘(25.4g,0.1mol)分三批加入上述体系,加完后室温搅拌10分钟,冷至0℃,将实施例1所得粗品用150ml二氯甲烷溶解30分钟内滴入上述体系,保持0℃继续搅拌1h,升至室温搅拌1.5h,加入200ml水,缓慢加入1m盐酸,将ph值调4,分液,有机相浓缩至约80ml,硅胶柱层析(乙酸乙酯:石油醚3:1)得产物化合物3(24.5g,0.078mol),实施例1和2的两步收率78%。
实施例3化合物6的制备
将锌粉(64g,1mol)和196mln,n-二甲基甲酰胺加入反应瓶中,氮气置换三次。加入1,2-二溴乙烷(13.9g,0.074mol),将反应液升温到85±2℃,保温十分钟。冰浴冷却到25±2℃,加入三甲基氯硅烷(4.8g,0.044mol),保温25±2℃10分钟。将反应液冷却到10~15℃,减压抽除易挥发物质。将实施例2制备的化合物3(24.5g,0.078mol)溶于dmf(196ml)配成溶液。控制温度在20+2℃下将该溶液滴加至反应液中。滴加完毕后搅拌2小时,制备锌试剂冷藏备用。
在四口瓶中依次加入2,5-二甲基-4-氰基碘苯(20.6g,0.08mol),p(o-tol)3(三(邻甲基)苯基磷)(0.23g,0.008mol)和pd2(dba)3(0.34g,0.004mol)溶入(128ml)dmf,氮气置换三次。将反应液加热到80~85℃。将制备好的锌试剂于2小时内滴加至反应体系中,滴加过程中温度逐渐上升到95+2℃,滴加完毕后保温反应半个小时,检测无原料剩余时停止反应,减压蒸馏除去溶剂,硅胶柱层析(乙酸乙酯:石油醚4:1)得产物(20.2g,0.064mol)两步收率81%。
实施例4化合物7的制备
向反应容器中加入实施例3制备的20.2g化合物6和320mlph7.2的磷酸盐缓冲液,搅拌均匀后加入尚科生物医药(上海)有限公司生产销售的酰胺水解酶(amd)2g,30℃下磁力搅拌反应,同时tlc检测反应进程至反应完毕后,将反应液快速升温至100℃,保持100秒后,立即冰水浴降温至30℃,然后投入尚科生物医药(上海)有限公司生产销售的腈水合酶(nht)1.5g,30℃搅拌反应,tlc检测反应完毕后硅藻土过滤反应液,液相采用有机相萃取三次,合并有机相,无水硫酸钠干燥,减压旋干,即得产品18.5g,两步酶催化合计收率97.1%。所制备产物的hplc分析谱图如附图1、手性hplc谱图如附图2,检测结果显示实施例4所制备的化合物7纯度接近100%,ee值计算结果为100%。
实施例5化合物2的制备
将化合物1(110g,0.5mol)溶于甲醇1l,将氨气通入上述混合液直至饱和,置于聚四氟乙烯内衬的闷罐加热到110℃反应12h,检测反应进行完全后停止加热,冷却至室温,将反应液减压除去溶剂,所得产物直接投下一步,无需进一步纯化。
实施例6化合物3的制备
将三苯基磷(262g,1mol),咪唑(68g,1mol)溶于3.5l二氯甲烷,将碘(190g,0.75mol)分三批加入上述体系,加完后室温搅拌10分钟,冷至0℃,将实施例5所得粗品用2000ml二氯甲烷溶解50分钟内滴入上述体系,保持0℃继续搅拌1h,升至室温搅拌1.5h,加入200ml水,缓慢加入1m盐酸,将ph值调4,分液,有机相浓缩至约300ml,硅胶柱层析(乙酸乙酯:石油醚3:1)得产物化合物3(120g,0.384mol),实施例5和6的两步收率76.7%。
实施例7化合物6的制备
将锌粉(640g,10mol)和2000mln,n-二甲基甲酰胺加入反应瓶中,氮气保护下加入1,2-二溴乙烷(111.2g,0.6mol),将反应液升温到90℃,保温十分钟。冰浴冷却到25℃,加入三甲基氯硅烷(38.4g,0.352mol),保温25±2℃10分钟。将反应液冷却到10~15℃,减压抽除易挥发物质。将实施例6制备的化合物3(120g,0.384mol)溶于dmf(1000ml)配成溶液。控制温度在20℃下将该溶液滴加至反应液中。滴加完毕后搅拌2小时,制备锌试剂冷藏备用。
在四口瓶中依次加入2,5-二甲基-4-氰基碘苯(100g,0.4mol),p(o-tol)3(三(邻甲基)苯基磷)(1g,0.035mol)和pd2(dba)3(2.1g,0.024mol)溶入(750ml)dmf,氮气置换三次。将反应液加热到95℃。将制备好的锌试剂于2小时内滴加至反应体系中,滴加过程中温度逐渐上升到95℃,滴加完毕后保温反应半个小时,检测无原料剩余时停止反应,减压蒸馏除去溶剂,硅胶柱层析(乙酸乙酯:石油醚4:1)得产物(96.8g,0.31mol)两步收率80%。
实施例8化合物7的制备
向反应容器中加入实施例7制备的96.8g化合物6和1600mlph7.0的磷酸盐缓冲液,搅拌均匀后加入尚科生物医药(上海)有限公司生产销售的酰胺水解酶(amd)8g,30℃下磁力搅拌反应,同时tlc检测反应进程至反应完毕后,将反应液快速升温至100℃,保持180秒后,立即冰水浴降温至20℃,然后投入尚科生物医药(上海)有限公司生产销售的腈水合酶(nht)6g,30℃搅拌反应,tlc检测反应完毕后硅藻土过滤反应液,液相采用有机相萃取三次,合并有机相,无水硫酸钠干燥,减压旋干,即得产品90g,两步酶催化合计收率94.5%。所制备化合物7的图谱检测结果同实施例4。
- 还没有人留言评论。精彩留言会获得点赞!
- 一类以ent-贝叶烷型二萜为手性臂的分子钳化合物及其制备方法和应用
- 含有ent-贝叶烷骨架的手性冠醚及其制备方法和应用
- 含有ent-贝叶烷骨架的开链手性冠醚及其制备和应用
- 制备分子印迹过氧化聚吡咯/纳米金修饰电极及其应用于电化学识别半胱氨酸对映体的制作方法
- 一类以(1s,2s)-1,2-环己二胺为隔离基、以异斯特维醇为手性臂的分子钳化合物及其制 ...的制作方法
- 一种l-脯氨酸类有机小分子催化剂及其制备和应用
- 一种d-乳酸脱色的新工艺的制作方法
- 钻孔用辅导片的感旋光性涂覆树脂及其应用
- 手性nd-322,nd-364或nd-364亚砜类似物及其制备方法
- 基于壳聚糖/海藻酸钠的手性传感器对含有锌离子的色氨酸对映体的手性识别的制作方法