一种组蛋白去乙酰化酶和微管双靶点抑制剂及其制备方法与流程
本发明属于组蛋白去乙酰化酶抑制剂合成
技术领域:
,具体地说,涉及一种组蛋白去乙酰化酶和微管双靶点抑制剂及其制备方法。
背景技术:
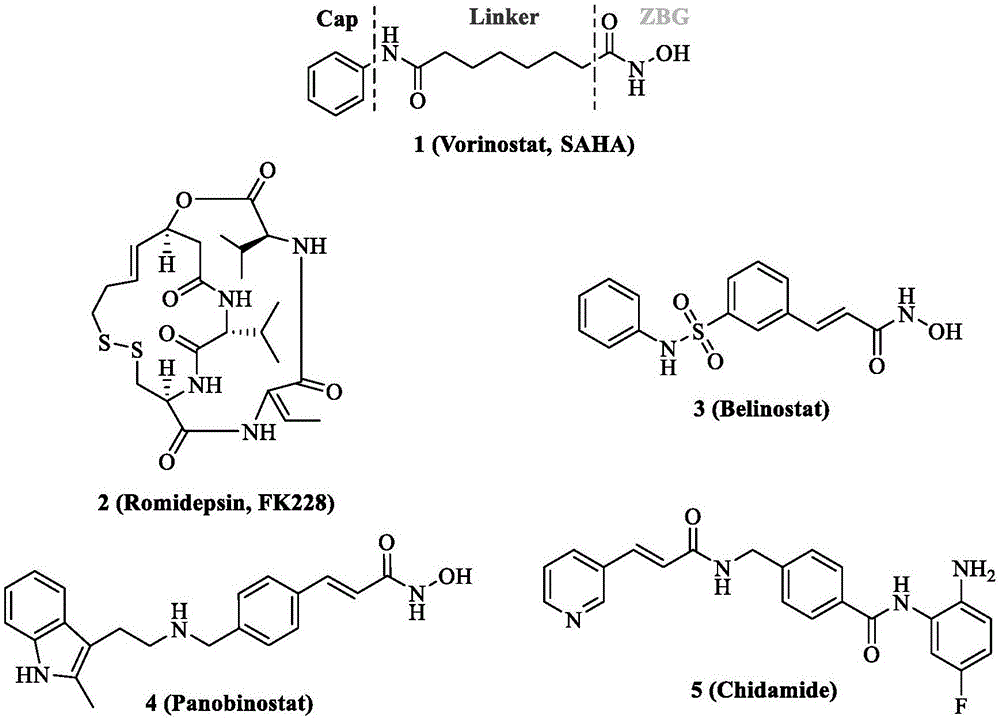
:组蛋白去乙酰化酶(histonedeacetylase,hdac)是维持染色体的基本组成单位核小体中组蛋白乙酰化状态平衡的关键酶类之一,其催化组蛋白的去乙酰化作用,与基因转录抑制密切相关,牵扯到促基因沉默的诸多过程,组蛋白及非组蛋白乙酰化状态失衡与肿瘤的发生、发展密切相关,因此组蛋白去乙酰化酶也是抗肿瘤药物设计中的热门靶标。组蛋白去乙酰化酶抑制剂(histonedeacetylaseinhibitor,hdaci)是一类化合物,有干扰组蛋白去乙酰化酶的功能。hdacis通过增加细胞内组蛋白的乙酰化程度,提高p21等基因的表达水平等途径,抑制肿瘤细胞的增殖,诱导肿瘤细胞分化和(或)凋亡。对肿瘤细胞迁移、侵袭、转移的抑制作用和抗肿瘤血管生成作用也被证实,因此hdaci的开发已成为肿瘤靶向治疗的研究热点。hdaci的药效团由三部分组成,cap结构,用于识别hdac活性口袋的入口处;锌离子螯合基团(zbg),用于螯合hdac催化口袋底部的锌离子;连接链,用于连接cap与zbg,并与活性口袋中的疏水性通道相互作用。而根据结构特点,已报道的hdaci可大致分为4个大类:异羟肟酸类、苯甲酰胺类、环肽类和短链脂肪酸类。其中异羟肟酸类包括曲古抑菌素和saha及其衍生物等。这一类hdac抑制剂的研究倍受瞩目,它被证明可在小剂量、低浓度情况下导致肿瘤分化,并选择性地抑制肿瘤生长而对正常细胞无毒副作用,更适用于临床。目前已有5种hdacis被批准上市用于临床(如图1所示)。现已上市的hdacs对实体瘤的治疗效果不理想以及伴随多种副作用等,这严重限制了hdaci在临床上的应用范围。为了解决这一难题,双靶点的hdac抑制剂和设计与合成提供了一种可行的方案。由于肿瘤的发生和发展涉及多个信号传导通路。仅仅抑制一条通路难以完全抑制肿瘤的增殖。而研究表明,多靶点抗癌药物可提高单靶点抗癌药物的治疗效果,并降低耐药性,是抗癌药物研发的重要研究方向。组蛋白去乙酰化酶与肿瘤的发生密切相关,其抑制剂可以降低肿瘤细胞凋亡的阈值,具有广泛的抗肿瘤活性,并且可与多种抗肿瘤药物联合使用发挥协同作用。这为双靶点hdac抑制剂的设计与开发提供了理论基础。目前已有两个多靶点hdac抑制剂处于临床研究阶段。而其他的hdac多靶点抑制剂如impdh-hdac双靶点抑制剂,topoⅱ-hdac双靶点抑制剂,egfr/her2-hdac双靶点抑制剂和hmga-hdac双靶点抑制剂等等也都表现出了较好的抗肿瘤活性。微管作为重要的细胞骨架,在细胞中起支撑作用,在细胞分裂前期,微管解聚重组成纺锤体,同时微管还参与细胞的信号传导和迁移。抑制微管被证明可阻滞肿瘤细胞有丝分裂和周期进程,诱导肿瘤细胞凋亡,破坏肿瘤血管并抑制血管生成。因此微管被认为是抗肿瘤药物研发的重要靶点。研究表明,秋水仙碱具有抑制微管蛋白聚合的作用。把秋水仙碱和hdac抑制剂联合使用时,两者具有协同抗肿瘤作用。受此启发,zhang等通过在秋水仙碱分子中引入saha的尾部基团作为hdac抑制剂的锌离子螯合基团,合成了一系列新颖的化合物。研究结果表明这些化合物不仅具有hdac抑制作用,同时,这些化合物对bel-7401细胞周期的g2/m期有明显延长作用,表明此类化合物也有抑制微管蛋白聚合的作用。在mtt实验中发现这类化合物对人表皮a431细胞、人肺腺癌a549细胞、结肠癌hct-116细胞、乳腺癌mcf-7细胞、前列腺癌pc-3细胞都有抑制活性。查尔酮sd400能够有效的抑制微管蛋白的组装,具有抗肿瘤的活性。研究人员已经合成了一系列的查尔酮类似物,实验结果表明这些类似物具有强烈的细胞毒性,能够有效地抑制肿瘤的增殖,其中化合物2a和2b为已报到的微管聚合抑制剂(如图2所示),两个化合物对多种肿瘤的ic50值达到了纳摩尔的级别。尽管微管是一种重要的抗肿瘤药物研发靶点,但微管与hdac双靶点抑制剂的报道极少。技术实现要素:有鉴于此,本发明提供了一种组蛋白去乙酰化酶和微管双靶点抑制剂及其制备方法与应用。为了解决上述技术问题,本发明公开了一种组蛋白去乙酰化酶和微管双靶点抑制剂,如i)或ii)所示:可选地,所述(i)的具体结构及名称如下所示:1)名称为n-羟基-7-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-庚酰胺,简称wbl-1,其化学结构如下所示:2)名称为n-羟基-8-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-辛酰胺,简称wbl-2,其化学结构如下所示:3)名称为n-羟基-7-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-庚酰胺,简称wbl-3,其化学结构如下所示:4)名称为n-羟基-8-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-辛酰胺,简称wbl-4,其化学结构如下所示:所述(ii)的具体结构及名称如下所示:1)名称为n1-羟基-n7-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-庚二酰胺,简称wbl-5,其化学结构如下所示:2)名称为n1-羟基-n8-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-辛二酰胺,简称wbl-6,其化学结构如下所示:3)名称为n1-羟基-n7-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-庚二酰胺,简称wbl-7,其化学结构如下所示:4)名称为n1-羟基-n8-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-辛二酰胺,简称wbl-8,其化学结构如下所示:本发明还公开了一种组蛋白去乙酰化酶和微管双靶点抑制剂的制备方法,包括以下步骤:步骤1、合成a或b:称取3’,4’,5’-三甲氧基苯乙酮或3’,4’,5’-三甲氧基苯丙酮与3-羟基-4-甲氧基苯甲醛溶于甲醇溶剂中,将50%(w/v)的氢氧化钠溶液加入到上述混合物中,混合物在氮气保护下过夜;加盐酸调节混合物的ph至1,二氯甲烷萃取两次,tlc检查反应情况;无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体a或b;步骤2、合成ames或bmes:将a或b与m-溴x酸甲酯溶于dmf,加入两倍量的无水碳酸钾,60℃油浴过夜,tlc检查;加水与乙酸乙酯萃取,无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体ames或bmes;步骤3、合成amac或bmac:将ames或bmes与1n氢氧化锂混合溶于体积比1:1的水与thf中;25℃油浴过夜,tlc检查反应情况,调ph为酸性,加ea萃取,无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体amac或bmac;步骤4、合成amthp或bmthp:将amac或bmac、dcc和nh2othp溶于dcm,25℃油浴过夜,tlc检查反应情况;过滤出去dcc浓缩后硅胶柱色谱法纯化得到黄色固体amthp或bmthp;步骤5、合成(i):将amthp或bmthp和csa溶于甲醇,68℃油浴3h,tlc检查反应情况,其中,dcm:meoh=20:1;hcl调至酸性,加水,ea萃取无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体(i)。可选地,所述步骤1中的3’,4’,5’-三甲氧基苯乙酮或3’,4’,5’-三甲氧基苯丙酮与3-羟基-4-甲氧基苯甲醛的摩尔比为1:1,tlc检查中的pe:ea=2:1;硅胶柱色谱法纯化中的pe:ea=4:1;所述步骤2中的a或b与m-溴x酸甲酯的摩尔量比例为1:1.1,m为7或8;x为庚或辛;tlc检查中的pe:ea=2:1;硅胶柱色谱法纯化中的pe:ea=6:1。可选地,所述步骤3中的ames或bmes与1n氢氧化锂的摩尔比例为1:1.5;tlc检查中的pe:ea=1:1;硅胶柱色谱法纯化中的pe:ea=1:1;所述步骤4中的amac或bmac:dcc:nh2othp=1mol:1.2mol:1.2mol;tlc检查中的dcm:ea=3:1;硅胶柱色谱法纯化中的dcm:ea=8:1。可选地,所述步骤5中的amthp或bmthp:csa=4.8mol:1mol;硅胶柱色谱法纯化中的dcm:meoh=100:1。本发明还公开了一种组蛋白去乙酰化酶和微管双靶点抑制剂的制备方法,包括以下步骤:步骤1、合成c或e:称取3’,4’,5’-三甲氧基苯乙酮或3’,4’,5’-三甲氧基苯丙酮和3-羟基-4-硝基苯甲醛溶于甲醇溶剂中,将50%(w/v)的氢氧化钾溶液加入到上述混合物中,混合物在氮气保护下过夜。tcl检查反应情况,反应完成后,加盐酸调节混合物的ph至1,二氯甲烷萃取两次;无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体c或e;步骤2、合成d或f:硝基化合物c或e悬浮于100毫升乙醇,加入10毫升蒸馏水,再加入还原铁粉和氯化铵;该混合物再90℃下回流搅拌4小时;降至室温,真空条件下除掉乙醇,再加入100毫升蒸馏水,将ph值用0.1m氢氧化钠水溶液调至弱碱性(8-9);用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥;粗品过硅胶柱纯化;得到黄色纯品d或f;步骤3、合成cmes或dmes:氨基化合物d或f溶于10毫升无水dmf,加入一倍量的x二酸单甲酯,搅拌下加入两倍量hatu和四倍量的dipea;混合物于室温下搅拌过夜,加入饱和食盐水,用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥,粗品使用柱层析纯化,得到纯品cmes或dmes;步骤4、合成cmac或dmac:cmes或dmes与1n氢氧化锂溶于体积比1:1的水与thf中;25℃油浴过夜,tcl检查反应进程,调ph为酸性,加ea萃取,无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到黄色固体cmac或dmac:;步骤5、合成cmthp或dmthp:将cmac或dmac、dcc和nh2othp溶于dcm,25℃油浴过夜,tcl检查反应情况;过滤出去dcc浓缩后硅胶柱色谱法纯化得到黄色固体cmthp或dmthp;步骤6、合成(ii):cmthp或dmthp(0.9g,1.6mmol):csa=4.8:1mol,溶于甲醇,68℃油浴3h,tcl检查反应情况;hcl调至酸性,加水,ea萃取无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化得到棕色固体。可选地,所述步骤1中的3’,4’,5’-三甲氧基苯乙酮与3-羟基-4-甲氧基苯甲醛的摩尔比为1:1,tlc检查中的pe:ea=2:1;硅胶柱色谱法纯化中的pe:ea=4:1;所述步骤2中的硝基化合物、还原铁粉和氯化铵的摩尔比为1:1:5;硅胶柱纯化的流动相为乙酸乙酯:石油醚=1:2。可选地,所述步骤3中的m为7或8,x为庚或辛;硅胶柱纯化的流动相为乙酸乙酯:石油醚=1:3;步骤4中的cmes或dmes与1n氢氧化锂的摩尔比例为1:1.5;tlc检查中的pe:ea=1:1;硅胶柱色谱法纯化中的pe:ea=1:1。可选地,所述步骤5中的cmac或dmac:dcc:nh2othp=1mol:1.2mol:1.2mol;tlc检查中的dcm:ea=3:1;硅胶柱色谱法纯化中的dcm:ea=8:1;所述步骤6中的cmthp或dmthp:csa=4.8mol:1mol;tlc检查中的dcm:meoh=20:1;硅胶柱色谱法纯化中的dcm:meoh=100:1。与现有技术相比,本发明可以获得包括以下技术效果:1)本发明首次合成了具有查尔酮骨架结构的新型组蛋白去乙酰化酶抑制剂。2)本发明设计并合成的化合物还具有微管聚合抑制活性。3)其中化合物wbl-1,wbl-4,wbl-8对包括实体瘤在内的多种肿瘤细胞都有强的抑制作用。当然,实施本发明的任一产品并不一定需要同时达到以上所述的所有技术效果。附图说明此处所说明的附图用来提供对本发明的进一步理解,构成本发明的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:图1是本发明
背景技术:
中的已上市的5个hdac抑制剂;图2是本发明
背景技术:
中的化合物2a与2b的结构;图3是本发明化合物的单浓度抑制混合hdac酶的结果。具体实施方式以下将配合实施例来详细说明本发明的实施方式,藉此对本发明如何应用技术手段来解决技术问题并达成技术功效的实现过程能充分理解并据以实施。实施例1wbl-1的制备wbl-1的结构式为:其化学名为:n-羟基-7-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-庚酰胺。其合成路线为:其合成方法的具体步骤如下:步骤1、合成a:称取3’,4’,5,-三甲氧基苯乙酮(21.02g,1mmol)和3-羟基-4-甲氧基苯甲醛(15.22g,1mmol)溶于200ml甲醇中,将50%(w/v)的氢氧化钠溶液160ml加入到上述混合物中,混合物在氮气保护下室温搅拌过夜。蒸除甲醇,并加1m盐酸调节混合物的ph值至1-2,用二氯甲烷20毫升萃取三次,收集二氯甲烷相并用无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=4:1)得到纯品a为黄色固体,产率52%,测熔点122℃;步骤2、合成a7es:a(3.4g,10mmol)与7-溴庚酸甲酯的摩尔量比例为1:1.1,两者溶于20ml无水n’,n’二甲基甲酰胺,加入两倍量的无水碳酸钾,60℃油浴加热搅拌过夜,tlc检测(pe:ea=2:1(v/v))至反应完全。加200ml水后用乙酸乙酯20毫升萃取3次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=6:1)得到a7es纯品为黄色固体,产率为67%;步骤3、合成a7ac:a7es(3.2g,6.7mmol)与1m氢氧化锂水溶液的摩尔比例为1:1.5,混合物溶于30毫升体积比1:2的水与四氢呋喃的混合溶剂中。25℃室温搅拌过夜,tlc检查反应情况(pe:ea=1:1(v/v)),蒸除四氢呋喃,调ph值为1-2,加乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(pe:ea=1:1)得到a7ac纯品为黄色固体,产率为68%。步骤4、合成a7thp:a7ac(2.2g,4.5mmol)溶于无水二氯甲烷,先后加入9mmol的二环己基碳二亚胺和9mmol的nh2othp,25℃室温搅拌过夜,tlc检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤除去反应残渣后,浓缩反应液,直接过硅胶柱纯化(二氯甲烷:乙酸乙酯=8:1)得到a7thp纯品为黄色固体,产率为59%。步骤5、合成wbl-1:a7thp(1.5g,2.7mmol):樟脑酸=4.8:1(mol/mol),溶于20ml甲醇中,68℃油浴搅拌回流3个小时,tlc检查反应情况(二氯甲烷:甲醇=20:1(v/v))。降至室温后,蒸除甲醇并加水,乙酸乙酯萃取,乙酸乙酯相用无水硫酸钠干燥,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-1纯品为黄色固体,产率为22%。产品wbl-1为黄色固体,1hnmr(500mhz,dmso)δ10.33(s,1h),8.65(s,1h),7.73(q,j=15.5hz,2h),7.48(d,j=1.8hz,1h),7.45(dd,j=8.4,1.8hz,1h),7.39(s,2h),7.02(d,j=8.4hz,1h),4.03(t,j=6.5hz,2h),3.89(s,6h),3.82(s,3h),3.76(s,3h),1.95(t,j=7.3hz,2h),1.77–1.65(m,2h),1.56–1.47(m,2h),1.47–1.37(m,2h),1.31(m,2h).13cnmr(126mhz,dmso)δ188.42,169.56,153.32,151.92,148.77,144.85,142.28,133.79,128.00,123.81,120.13,113.38,112.27,106.66,68.85,60.64,56.69,56.12,32.70,29.12,28.82,25.78,25.56.[m+h]+=487.2208.实施例2wbl-2的制备wbl-2的化学式为:其化学名称为:n-羟基-8-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-辛酰胺。其合成路线为:其合成方法的具体步骤如下:步骤1、合成a:同实施例1步骤1;步骤2、合成a8es:a(3.44g,10mmol)与8-溴辛酸甲酯的摩尔量比例为1:1.1,两者溶于20ml无水n’,n’二甲基甲酰胺,加入两倍量的无水碳酸钾,60℃油浴过夜,tlc检查反映情况(石油醚:乙酸乙酯=2:1(v/v))。加水与乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=6:1)得到黄色固体,产率73%。步骤3、合成a8ac:a8es(3.6g,7.3mmol)与1m氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与四氢呋喃混合溶液中。25℃室温搅拌过夜,tlc检查反应情况(石油醚:乙酸乙酯=1:1),调ph为1-2,加20ml乙酸乙酯萃取3次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=1:1)得到a8ac纯品为黄色固体,产率为63%。步骤4、合成a8thp:a8ac(2.3g,4.9mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷中,25℃室温搅拌过夜,tlc检查反应情况dcm:ea=3:1(v/v);过滤出去反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到a8thp纯品为黄色固体,产率为54%;步骤5、合成wbl-2:a8thp(1.5g,2.6mmol):樟脑酸=4.8:1(mol/mol),溶于15毫升甲醇,68℃油浴搅拌3小时,tlc检查反应情况(二氯甲烷:甲醇=20:1(v/v))。加水,乙酸乙酯萃取3次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-2纯品为黄色固体,产率为25%。产品wbl-2为黄色固体,1hnmr(500mhz,dmso)δ10.32(s,1h),8.64(s,1h),7.73(q,j=15.5hz,2h),7.49(d,j=2hz,1h),7.45(dd,j=10.5,2hz,1h),7.40(s,2h),7.03(d,j=8.4hz,1h),4.04(t,j=8.1hz,2h),3.90(s,6h),3.82(s,3h),3.76(s,3h),1.94(t,j=9.2hz,2h),1.72(m,2h),1.50(m,2h),1.41(m,2h),1.33(m,2h),1.26(m,2h).13cnmr(126mhz,dmso)δ188.42,169.58,153.32,151.91,148.78,144.85,142.28,133.79,128.00,123.80,120.11,113.31,112.25,106.65,68.86,60.64,56.68,56.11,32.71,29.16,29.01,28.93,25.94,25.54.[m+h]+=501.2366.实施例3wbl-3的制备wbl-3的化学式为:其化学名称为:n-羟基-7-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-庚酰胺。其合成路线为:其合成方法的具体步骤如下:步骤1、合成b:称取摩尔量相当的3’,4’,5’-三甲氧基苯丙酮(15g,66mmol)和3-羟基-4-甲氧基苯甲醛(33g,66mmol)溶于甲醇溶剂中,将11ml50%(w/v)的氢氧化钠溶液加入到上述混合物中,混合物在氮气保护下搅拌反应过夜,tlc检查反应情况(石油醚:乙酸乙酯=2:1(v/v))。加1m盐酸调节混合物的ph至1-2,100ml二氯甲烷萃取两次。无水硫酸钠干燥二氯甲烷相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=4:1)得到棕黄色纯品固体,产率64%;步骤2、合成b7es:b(3.5g,10mmol)与7-溴庚酸甲酯的摩尔量比例为1:1.1,两者溶于无水n,n-二甲基甲酰胺,加入两倍摩尔量的无水碳酸钾,60℃油浴搅拌过夜,tlc检查(石油醚:乙酸乙酯=2:1(v/v))。加水与乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=6:1)得到b7es纯品为黄色固体,产率76%;步骤3、合成b7ac:b7es(3.7g,7.5mmol)与1n氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与thf中;25℃室温搅拌过夜,tlc检查反应情况(pe:ea=1:1),调ph值至1-2,加30ml乙酸乙酯萃取三次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(p石油醚:乙酸乙酯=1:1)得到b7ac纯品为黄色固体,产率54%;步骤4、合成b7thp:b7ac(2.0g,4mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷,25℃室温搅拌过夜,tlc检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤出去反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到b7thp纯品为黄色固体,产率51%;步骤5、合成wbl-3:b7thp(1.2g,2mmol):樟脑酸=4.8:1(mol/mol),溶于甲醇,68℃油浴搅拌反应3小时,tlc检查反应情况(二氯甲烷:甲醇=20:1(v/v))。加水,乙酸乙酯萃取三次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-3纯品为棕色固体,产率9%。产品wbl-3为黄色固体,1hnmr(500mhz,dmso)δ10.32(s,1h),8.64(s,1h),7.19–7.07(m,3h),7.03(d,j=8.3hz,1h),6.97(s,1h),3.96(t,j=6.4hz,2h),3.81(s,6h),3.80(s,3h),3.76(s,3h),2.19(s,3h),1.94(t,j=7.3hz,2h),1.70(m,2h),1.50(m,2h),1.40(m,2h),1.29(m,2h).13cnmr(126mhz,dmso)δ197.89,169.54,152.94,150.13,148.30,141.89,140.98,134.21,133.89,128.46,123.74,115.25,112.24,107.27,68.63,60.58,56.48,56.05,32.68,29.09,28.81,25.73,25.54,15.04.[m+h]+=501.2364.实施例4wbl-4的制备其化学式为:其化学名称为:n-羟基-8-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯氧基-辛酰胺;其合成路线为:其合成方法的具体步骤如下:步骤1、合成b:同实施例3;步骤2、合成b8es:b(3.5g,10mmol)与8-溴辛酸甲酯的摩尔量比例为1:1.1,两者溶于无水n,n-二甲基甲酰胺,加入两倍摩尔量的无水碳酸钾,60℃油浴搅拌过夜,tlc检查(石油醚:乙酸乙酯=2:1(v/v));加水与乙酸乙酯萃取,乙酸乙酯相用无水硫酸钠干燥,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=6:1)得到b8es纯品为黄色固体,产率82%;步骤3、合成b8ac:b8es(4g,8mmol)与1m氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与四氢呋喃中;25℃室温搅拌过夜,tlc检查反应情况(石油醚:乙酸乙酯=1:1(v/v)),调ph值至1-2,加乙酸乙酯萃取3次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=1:1)得到b8es纯品为黄色固体,产率45%;步骤4、合成b8thp:b8ac(1.7g,3.5mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷,25℃室温搅拌过夜,tlc检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤出去dcc反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到b8thp纯品为黄色固体,产率88%;步骤5、合成wbl-4:b8thp(1.8g,3mmol):樟脑酸=4.8:1(mol/mol),溶于甲醇,68℃油浴搅拌3小时,tlc检查反应情况(二氯甲烷:甲醇=20:1)。使用1mhcl将ph值调至1-2,加入水,乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-10纯品为棕色固体,产率12%。产品wbl-4为黄色固体,1hnmr(500mhz,dmso)δ10.31(s,1h),8.64(s,1h),7.15-7.11(m,3h),7.03(d,j=8.3hz,1h),6.97(s,1h),3.96(t,j=6.4hz,2h),3.82(s,6h),3.80(s,3h),3.76(s,3h),2.19(s,3h),1.94(t,j=7.3hz,2h),1.69(m,2h),1.49(m,2h),1.39(m,2h),1.42–1.21(m,6h).13cnmr(126mhz,dmso)δ197.89,169.55,152.94,150.13,148.31,141.89,140.98,134.21,133.89,128.46,123.72,115.25,112.24,107.27,68.66,60.58,56.49,56.05,32.70,29.13,28.99,28.92,25.88,25.52,15.04.[m+h]+=515.2520.实施例5wbl-5的制备wbl-5的化学式为:其化学名称为:n1-羟基-n7-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-庚二酰胺;其合成路线为:其合成方法的具体步骤如下:步骤1、合成c:称取摩尔量相当的3’,4’,5’-三甲氧基苯乙酮(21g,100mmol)和3-羟基-4-硝基苯甲醛(15g,100mmol)溶于200ml甲醇中,将8ml50%(w/v)的氢氧化钠溶液加入到上述混合物中,混合物在氮气保护下过夜。tcl检查反应情况(石油醚:乙酸乙酯=2:1(v/v)),反应完成后,加1mhcl调节混合物的ph至1-2,100ml二氯甲烷萃取两次。无水硫酸钠干燥二氯甲烷相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=4:1)得到c的纯品为黄色固体,产率62%;步骤2、合成d:硝基化合物c(18g,100mmol)悬浮于200ml乙醇,加入20ml蒸馏水,再加入还原铁粉(28g,500mmol)和氯化铵(26g,500mmol);该混合物再90℃下回流搅拌4小时;待降至室温,真空条件下蒸除乙醇,再加入200ml蒸馏水,将ph值用0.1m氢氧化钠水溶液调至弱碱性(8-9);用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥。粗品过硅胶柱纯化,流动相为乙酸乙酯:石油醚=1:2。得到黄色纯品d,产率78%;步骤3、合成c7es:氨基化合物(2.5g,7.5mmol)溶于10毫升无水n,n-二甲基甲酰胺,加入1.2当量的庚二酸单甲酯,搅拌下加入两倍量hatu和四倍量的n,n-二异丙基乙胺;混合物于室温下搅拌过夜,加入饱和食盐水,用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥,粗品使用柱层析纯化,流动相为乙酸乙酯:石油醚=1:3,得到纯品,产率分别为73%;步骤4、合成c7ac:c7es(2.7g,5.4mmol)与1m氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与四氢呋喃中;25℃室温搅拌过夜,tcl检查反应进程(石油醚:乙酸乙酯=1:1(v/v)),调ph为酸性,加乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=1:1)得到c7ac纯品为黄色固体,产率为61%;步骤5、合成c7thp:c7ac(1.5g,3.2mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷,25℃室温搅拌过夜,tcl检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤出去反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到c7thp纯品为黄色固体,产率50%;步骤6、合成wbl-5:c7thp(0.9g,1.6mmol):csa=4.8:1mol,溶于甲醇,68℃油浴3h,tcl检查反应情况(dcm:meoh=20:1)。用1mhcl将溶液调至酸性,加水,乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-5纯品为棕色固体,产率17%。产品wbl-5为棕黄色固体,1hnmr(500mhz,dmso)δ10.35(s,1h),8.66(s,1h),7.72(q,j=15.5hz,2h),7.52(d,j=2.0hz,1h),7.46(dd,j=8.4,2.0hz,1h),7.06(s,2h),6.98(d,j=8.4hz,1h),3.89(s,6h),3.82(s,3h),3.76(s,3h),2.36(t,j=7.3hz,2h),1.96(t,j=7.3hz,2h),1.62(m,2h),1.49(m,2h),1.29(m,2h).13cnmr(126mhz,dmso)δ188.42,179.81,169.56,153.32,153.92,138.77,134.85,130.28,128.79,125.80,121.81,119.13,112.38,112.27,100.66,60.82,56.71,55.80,38.30,29.12,28.82,26.01,24.95.[m+h]+=501.2235.实施例6wbl-6的制备其化学式为:其化学名称为:n1-羟基-n8-(2-甲氧基-5-(3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-辛二酰胺;其合成路线为:其合成方法的具体步骤如下:步骤1、合成c:同实施例5;步骤2、合成d:同实施例5;步骤3、合成c8es:氨基化合物(2.5g,7.5mmol)溶于10ml无水n,n-二甲基甲酰胺,加入一倍量的辛二酸单甲酯,搅拌下加入两倍量hatu和四倍量的n,n-二异丙基乙胺。混合物于室温下搅拌过夜,加入饱和食盐水,用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥,粗品使用柱层析纯化,流动相为乙酸乙酯:石油醚=1:3,得到纯品c8es,产率分别为78%;步骤4、合成c8ac:c8es(2.9g,5.8mmol)与1m氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与四氢呋喃中。25℃室温搅拌过夜,tcl检查反应进程(石油醚:乙酸乙酯=1:1(v/v)),蒸除四氢呋喃后,调溶液ph为酸性,加乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=1:1)得到c8ac为黄色固体,产率为66%;步骤5、合成c8thp:c8ac(1.7g,3.8mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于50ml无水二氯甲烷,25℃室温搅拌过夜,tcl检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤去除反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到c8thp纯品为黄色固体,产率56%;步骤6、合成wbl-6:c8thp(1.2g,2.1mmol):樟脑酸=4.8:1(mol/mol),溶于甲醇,68℃油浴搅拌3小时,tcl检查反应情况(二氯甲烷:甲醇=20:1(v/v))。加水并蒸除甲醇,乙酸乙酯萃取三遍,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1),得到wbl-6为棕色固体,产率19%。产品wbl-6为棕色固体,1hnmr(500mhz,dmso)δ10.36(s,1h),8.67(s,1h),7.73(q,j=15.5hz,2h),7.52(d,j=2.0hz,1h),7.49(dd,j=8.4,2.0hz,1h),7.05(s,2h),6.97(d,j=8.4hz,1h),3.89(s,6h),3.82(s,3h),3.76(s,3h),2.37(t,j=7.3hz,2h),1.97(t,j=7.3hz,2h),1.63(m,2h),1.50(m,2h),1.31(m,2h),1.29(m,2h).13cnmr(126mhz,dmso)δ188.60,179.82,169.66,153.62,153.97,138.87,134.90,130.29,128.81,125.82,121.83,119.16,112.40,112.31,100.67,60.81,56.72,55.81,38.32,29.11,28.83,27.99,26.01,24.96.[m+h]+=515.2391.实施例7wbl-7的制备其化学式为:其化学名称为:n1-羟基-n7-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-庚二酰胺;其合成路线为:其合成方法的具体步骤如下:步骤1、合成e:称取摩尔量相当的3’,4’,5’-三甲氧基苯丙酮(22g,100mmol)和3-羟基-4-硝基苯甲醛(15g,100mmol)溶于甲醇溶剂中,将8g50%(w/v)的氢氧化钠溶液加入到上述混合物中,混合物在氮气保护下室温搅拌过夜。tcl检查反应情况(二氯甲烷:乙酸乙酯=2:1(v/v)),反应完成后,加1mhcl调节混合物的ph至1-2,200ml二氯甲烷萃取两次。无水硫酸钠干燥二氯甲烷相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=4:1)得到e纯品为黄色固体,产率65%;步骤2、合成f:硝基化合物e(18g,100mmol)悬浮于300毫升乙醇中,加入20毫升蒸馏水,再加入还原铁粉(28g,500mmol)和氯化铵(26g,500mmol);该混合物在90℃下回流搅拌4小时;降至室温后,真空条件下蒸除乙醇,再加入500毫升蒸馏水,将ph值用0.1m氢氧化钠水溶液调至弱碱性(8-9);用200ml乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥。粗品过硅胶柱纯化,流动相为乙酸乙酯:石油醚=1:2。得到f为黄色固体,产率58%;步骤3、合成d7es:氨基化合物f(2.5g,7.5mmol)溶于20毫升无水n,n-二甲基甲酰胺,加入一倍量的庚二酸单乙酯,搅拌下加入两倍量hatu和四倍量的n,n-二异丙基乙胺。混合物于室温下搅拌过夜,加入饱和食盐水,用20ml乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥,粗品使用柱层析纯化,流动相为乙酸乙酯:石油醚=1:3,得到纯品,产率分别为67%。步骤4、合成d7ac:d7es(2.5g,5mmol)与1m氢氧化锂的摩尔比例为1:1.5,混合物溶于30ml体积比1:2的水与四氢呋喃中。25℃室温搅拌过夜,tcl检查反应进程(石油醚:乙酸乙酯=1:1(v/v)),蒸除四氢呋喃并调溶液的ph至酸性,加乙酸乙酯萃取三次,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(石油醚:乙酸乙酯=1:1)得到d7ac纯品为黄色固体,产率为57%;步骤5、合成d7thp:d7ac(1.3g,2.8mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷,25℃室温搅拌过夜,tcl检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤出去反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到黄色固体,产率58%;步骤6、合成wbl-7:d7thp(0.9g,1.6mmol):樟脑酸=4.8:1(mol/mol),溶于甲醇,68℃油浴搅拌3小时,tcl检查反应情况(二氯甲烷:甲醇=20:1)。加水并蒸除甲醇,乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:甲醇=100:1)得到wbl-7纯品为棕色固体,产率13%。产品wbl-7为棕色固体,1hnmr(500mhz,dmso)δ10.39(s,1h),8.66(s,1h),7.61–7.49(m,3h),7.04(s,2h),6.97(d,j=8.4hz,1h),3.81(s,6h),3.80(s,3h),3.76(s,3h),2.35(t,j=7.3hz,2h),2.19(s,3h),1.96(t,j=7.3hz,2h),1.65(m,2h),1.51(m,2h),1.29(m,2h).13cnmr(126mhz,dmso)δ190.89,179.82,169.90,153.66,149.13,146.31,140.89,134.21,133.89,127.46,126.70,125.72,117.25,114.24,100.27,60.86,56.51,55.89,38.33,32.56,27.76,25.11,24.91,15.01.[m+h]+=515.2392.实施例8wbl-8的制备wbl-8的化学式为:其化学名称为:n1-羟基-n8-(2-甲氧基-5-(2-甲基-3-酮基-3-(3,4,5-三甲氧基苯基)-1-(e)-丙烯基)苯基-辛二酰胺;其合成路线为:其合成方法的具体步骤如下:步骤1、合成e:同实施例7;步骤2、合成f:同实施例7;步骤3、合成d8es:氨基化合物(2.5g,7.5mmol)溶于10毫升无水dmf,加入一倍量的丁二酸单甲酯,搅拌下加入两倍量hatu和四倍量的dipea。混合物于室温下搅拌过夜,加入饱和食盐水,用乙酸乙酯萃取三遍,乙酸乙酯层用无水硫酸钠干燥,粗品使用柱层析纯化,流动相为乙酸乙酯:石油醚=1:3,得到纯品,产率分别为78%;步骤4、合成d8ac:d8es(3.8g,7.2mmol)与1n氢氧化锂的摩尔比例为1:1.5,混合物溶于体积比1:1的水与thf中。25℃油浴过夜,tcl检查反应进程(pe:ea=1:1),调ph为酸性,加ea萃取,无水硫酸钠干燥滤液,浓缩后硅胶柱色谱法纯化(pe:ea=1:1)得到黄色固体,产率为50%;步骤5、合成d8thp:d8ac(1.8g,3.6mmol):二环己基碳二亚胺:nh2othp=1:1.2:1.2(mol/mol),溶于无水二氯甲烷,25℃室温搅拌过夜,tlc检查反应情况(二氯甲烷:乙酸乙酯=3:1(v/v))。过滤去除反应残渣,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=8:1)得到d8thp纯品为黄色固体,产率60%;步骤6、合成wbl-8:d8thp(1.3g,2.1mmol):樟脑磺酸=4.8:1(mol/mol),溶于甲醇,68℃油浴搅拌反应3小时,tlc检查反应情况(二氯甲烷:甲醇=20:1)。加水并蒸除甲醇,乙酸乙酯萃取,无水硫酸钠干燥乙酸乙酯相,浓缩后硅胶柱色谱法纯化(二氯甲烷:乙酸乙酯=100:1)得到wbl-8纯品为棕色固体,产率17%。产品wbl-8为棕色固体,1hnmr(500mhz,dmso)δ10.39(s,1h),8.66(s,1h),7.61–7.49(m,3h),7.04(s,2h),6.97(d,j=8.4hz,1h),3.81(s,6h),3.80(s,3h),3.76(s,3h),2.35(t,j=7.3hz,2h),2.19(s,3h),1.96(t,j=7.3hz,2h),1.65(m,2h),1.51(m,2h),1.29(m,2h).13cnmr(126mhz,dmso)δ190.90,179.83,169.88,153.67,149.12,146.32,140.90,134.22,133.90,127.45,126.71,125.72,117.26,114.25,100.30,60.85,56.52,55.88,38.34,32.58,27.86,27.76,25.12,24.92,15.02.[m+h]+=529.2550.下面结合具体的实验数据来说明本发明的技术效果:图3为8个化合物的单浓度抑制混合hdac酶的结果,化合物浓度皆为1μm。saha为阳性对照药,dmso为溶剂。该结果证明化合物wbl-1到wbl-8都初步表现出了对hdac混合酶的强效抑制活性,其中化合物wbl-4的对hdac混合酶的单农渡抑制率还略优于对照药物saha,wbl-8的活性与saha接近。表1:8个化合物对hdac1,6,8的抑制活性(nm)化合物名称hdac1hdac6hdac8wbl-125.811.2>1000wbl-250.717.1>1000wbl-3377.0130.7>1000wbl-436.114.2>1000wbl-536.822.6>1000wbl-656.927.2>1000wbl-7211.297.8>1000wbl-846.619.3>1000saha44.621.6540.6acy121573.08.0126.0其中,saha与acy1215为阳性对照药,单位为nm。表1中所列结果表明所选的8个化合物对hdac1和6两种亚型,特别是hdac6亚型具有很好的选择性,而对hdac8在1μm浓度下基本上没有活性。因此表明,8个化合物对hdac1和6两种亚型具有较好的选择性。表2:8个化合物对微管蛋白聚合的抑制活性(μm)其中,阳性对照为化合物2a,单位为μm。表2中所列结果表明在hdac酶抑制活性较好的8个化合物同样也表现出了对微管聚合的抑制活性。表38个化合物对3种肿瘤细胞增殖的抑制活性(μm)化合物hct-116(ic50)mda-mb-231(ic50)hl-60(ic50)saha5.065.152.882a3.637.561.91wbl-17.733.981.33wbl-2>10>105.63wbl-38.926.683.66wbl-47.443.831.13wbl-58.227.724.36wbl-69.065.564.89wbl-7>10>105.17wbl-87.764.161.68其中,阳性对照药为saha和2a,单位为μm。从上表3可以看出,化合物wbl-1,wbl-4和wbl-8三个化合物对mda-mb-231和hl-60两种癌细胞的抑制活性较好,且活性要比两种阳性药saha和2a要好一些,ic50值都低于5μm,所以这三种化合物都可以做潜在治疗癌症的候选药物。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种药物中间体2-芳酰基吲哚衍生物的合成方法
- 双重hdac/brd4抑制剂及其制备方法和应用
- 含有色氨酸基本骨架的邻苯二胺类选择性组蛋白去乙酰化酶抑制剂及其制备方法和应用
- 一种生产贝利司他中间体的工艺方法
- 组蛋白去乙酰化酶抑制剂治疗β亚科疱疹病毒的新用图
- 一种含有苯甘氨酸的肉桂酰胺类组蛋白去乙酰化酶抑制剂及其制备方法和应用
- 2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚-3-基)-n-羟基乙酰胺作为组蛋白去 ...的制作方法
- 一种组蛋白去乙酰酶抑制剂2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚-3-基-n ...的制作方法
- 一种组蛋白去乙酰酶抑制剂(e)-3-(2-(1-(4-氯苯甲酰基)-5-甲氧基-2-甲基-1氢-吲哚 ...的制作方法
- 小麦组蛋白去乙酰化酶基因TaHD2C及其应用