一种磺酸官能化的高分子嫁接碳纳米管的复合材料及其制备方法和应用与流程
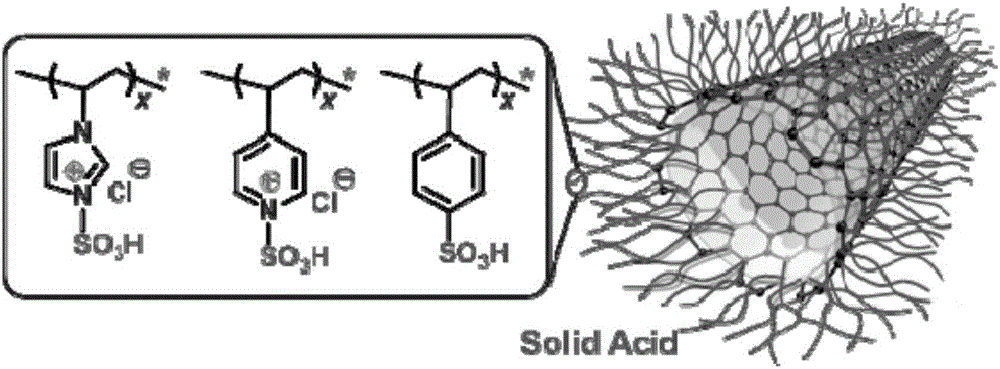
碳纳米管与磺酸官能化的高分子(P-SO3H)共价接枝得到的复合材料(CNT-P-SO3H),作为碳基材料的固体酸用于酯交换反应和酯化反应,表现出显著的催化性能。CNT-P-SO3H催化剂的杰出性质归功于介孔结构以及CNT外表面所包覆的P-SO3长链。这样的结构使得碳纳米管外表面密集的活性部位均匀分布。由此能促进反应物之间的接触以及利于试剂脱离催化剂。这些性质使得CNT-P-SO3H复合材料成为一种理想的碳材料固体酸用于酯交换反应和酯化反应。将磺酸化的高分子共价嫁接到碳纳米管上获得一系列聚合物—碳纳米管复合材料。包括聚(3-乙烯基-1-氯磺酸化咪唑)嫁接多壁纳米管(CNT-PVSAIC),聚(4-乙烯基-1-氯磺酸化吡啶)嫁接多壁碳纳米管(CNT-PVSAPC)以及聚苯乙烯磺酸嫁接多壁碳纳米管(CNT-PSSA)。为了提高碳纳米管的分散性和调节酸度,这种功能化技术易得各种碳纳米管表面的聚电解质刷。为潜在的催化应用,在碳纳米管上有选择性地引进官能团并密集地创造活性位。CNT-PVSAIC和CNT-PVSAPC都包含由磺酸基官能化的阳离子聚电解质链。但是,CNT-PSSA是由磺酸基嫁接的阴离子高分子刷组成。在三酸甘油酯与甲醇的酯交换反应以及油酸与甲醇的酯化反应中,所得的CNT-P-SO3H材料作为碳基固体酸在液相中表现出显著的催化性能。CNT-P-SO3H材料的优秀性能归结于介孔结构以及碳纳米管外表面包裹一层P-SO3H高分子,提供了密集但统一的活性位表面分布。此外,表面裸露的活性位有利于反应物的接触,并能促进试剂脱离催化剂。这些性能优点使得CNT-P-SO3H合成材料作为碳基固体酸有利于酯交换反应和酯化反应。
背景技术:
:生物柴油,提炼自动植物油,普遍用于拖拉机、卡车、船舶等。它是指以油料作物如大豆、油菜、棉、棕榈等,野生油料植物和工程微藻等水生植物油脂以及动物油脂、餐饮垃圾油等为原料油通过酯交换工艺制成的可代替石化柴油的再生性柴油燃料。生物柴油是生物质能的一种,它是生物质利用热裂解等技术得到的一种长链脂肪酸的单烷基酯。生物柴油是含氧量极高的复杂有机成分的混合物,这些混合物主要是一些分子量大的有机物,几乎包括所有种类的含氧有机物,如:醚、醛、酮、酚、有机酸、醇等。复合型生物柴油是以废弃的动植物油、废机油及炼油厂的副产品为原料,再加入催化剂,经专用设备和特殊工艺合成。作为一种不可再生石油柴油的替代物越来越引起关注。当前,工业生物柴油的生产主要是基于酯交换反应和酯化反应来获得脂肪酸甲酯。通过用碱催化剂或酸催化剂可以提高产率。使用固体酸催化剂易于简单的产品分离和催化剂的回收。此次之外,使用固体酸催化剂作为首选,是因为在石油原料中,三酸甘油酯的酯交换反应的同时促进游离脂肪酸的酯化反应且没有皂化反应。因此,在生物柴油生产过程中,固体酸能减少生产步骤。由此满足了催化生产生物柴油中绿色和可持续化学的要求。通过三酸甘油酯的酯交换反应和游离脂肪酸的酯化反应,各种固体酸催化剂已经用于脂肪酸甲酯的合成。磺酸基为无定型碳,被称为“糖催化剂”,因为他们疏水亲油的表面性质,促进有机试剂的扩散,在催化剂表面酸性-SO3H官能团的存在,报道称之为一种很有前景的酸性材料。但是,大多数磺化无定型碳比表面积极低,由此限制了反应物分子靠近酸性位点。此外,糖催化剂一般用发烟硫酸磺化部分碳化有机分子制得,在催化剂表面得到随机的,无定型的酸位。上述问题可以克服在拥有一个大的表面积的纳米结构材料的碳边缘-SO3H官能团的选择性沉积问题,从而促进大分子反应物向酸性位点的靠近。通过改进合成控制,一系列由碳纳米管,碳纳米纤维,石墨烯衍生出的固体酸拥有精细的结构,可调节的表面组成,纹理以及酸性。进来它们被开发成为碳基的酸性催化剂应用于各种反应。例如,聚(4-对苯乙烯磺酸)嫁接碳纳米管(CNT-PSSA)用于果糖脱水得到5-羟甲基糠醛和月桂酸和甲醇的酯化反应。报道指出,杂多酸与氮官能化的碳纳米管的杂化材料可用于乙酸乙酯的水解,环己酮肟的贝克曼重排反应以及甲苯与正辛烯的傅克烷基化反应。沉积在碳纳米管上的聚苯胺硫酸盐被开发用于三酸甘油酯的甲醇分解反应。苯磺酸官能化的碳纳米纤维用于三油酸甘油脂与甲醇的酯交换反应以及果糖脱水生成5-羟甲基糠醛的反应。磺化的石墨烯和磺化的氧化石墨烯可作用于乙酸乙酯的水解反应,乙酸乙酯与醇类的酯交换反应,木糖脱水成糠醛,碳水化合物转换成乙酰丙酸,羟甲基糠醛转换成生物燃料。技术实现要素:鉴于以上问题,本发明的目的是提供磺酸官能化的高分子嫁接碳纳米管的复合材料及其制备方法和应用。具体技术方案如下:一种磺酸官能化的高分子嫁接碳纳米管的复合材料,具有如下结构式:其中,Y表示可回收的聚(3-乙烯基-1-氯磺酸化咪唑)的聚合度,为5000~6000的整数,X表示无法回收的聚(3-乙烯基-1-氯磺酸化咪唑)的聚合度,为2000~3000;结构式左侧为多壁碳纳米管(CNT)。上述复合材料的制备方法,包括如下步骤:(1)称取40~60mg碳纳米管放入预先置有磁性搅拌子的干燥斯莱克瓶,抽真空,冲氮气,重复三次;(2)开大氮气的流率,分别量取200~300mLDMF(N,N-二甲基甲酰胺)和0.5~1.0mL1-乙烯基咪唑注入所述斯莱克瓶,将通氮气的所述斯莱克瓶放入超声仪中,超声0.5~1.5h;(3)超声完毕,加入5~10mg引发剂,在氮气条件下将装置放入65~75℃恒温油浴锅中搅拌反应40~50h;(4)反应完后,关闭氮气阀,从油浴锅中取出反应装置,冷却至室温;用DMF试剂稀释到400~600mL;离心分离,取下离心管壁上的黑色固体,用尼龙膜抽滤,并用DMF和无水乙醇冲洗,抽干;(5)从尼龙膜上取下黑色产物,于75~85℃真空干燥箱中烘干,得到PVI/CNT;(6)称取40~60mgPVI/CNT,200~300mL1,2-二氯乙烷以及0.1~0.15mL氯磺酸于斯莱克瓶中;室温下,搅拌反应1.5~2.5h,然后在60~70℃的恒温油浴锅中反应20~30h;反应完后,离心分离所得的固体产物CNT-PVSAIC,用二氯甲烷和无水乙醚冲洗,水泵抽干,75~85℃真空干燥箱中干燥20~30h。所述引发剂是AIBN。上述复合材料的应用,用于酯交换反应和酯化反应,表现出极好的催化活性,可降低工业生产生物柴油的成本,以及提高生物柴油的产率。所述酯交换反应是三酸甘油酯与甲醇的酯交换反应,所述酯化反应是油酸与甲醇的酯化反应。三酸甘油酯与甲醇的酯交换反应,过程如下:油酸与甲醇的酯化反应,过程如下:本发明的优点体现在:制备的固体酸催化剂用于三酸甘油酯与甲醇的酯交换反应和油酸与甲醇的酯化反应,表现出极好的催化活性,可降低工业生产生物柴油的成本,以及提高生物柴油的产率。附图说明图1是实施例1复合材料的结构示意图;图2是CNT-PVSAIC的分子结构图;图3是纯碳纳米管和CNT-P-SO3H复合材料的傅里叶变换红外光谱;图4是三种催化剂的氮气吸附-脱附等温曲线及对应的孔径分布曲线;图5是CNT-PVSAIC,CNT-PVSAPC及CNT-PSSA的TG曲线;图6是CNT-PVSAIC,CNT-PVSAPC,CNT-PSSA及CNT的XRD曲线;图7是CNT-PVSAIC,CNT-PVSAPC,CNT-PSSA及CNT的Raman曲线;图8是高分辨率透射电子显微镜图,其中:(a)、(b)、(c)分别是CNT-PVSAIC、CNT-PVSAPC和CNT-PSSA;图9是反应条件对产物影响的示意图;其中:(a)是CNT-PVSAIC催化三丁酸甘油酯与甲醇的酯交换反应的产物分布;(b)是温度对三丁酸甘油酯与甲醇的酯交换反应的影响;(c)是三丁酸甘油酯与甲醇的摩尔比对三丁酸甘油酯甲醇分解反应的影响;图10是催化剂循环回收示意图。具体实施方式下面,用实施例来进一步说明本
发明内容,但本发明的保护范围并不仅限于实施例。对本领域的技术人员在不背离本发明精神和保护范围的情况下做出的其它的变化和修改,仍包括在本发明保护范围之内。实施例1傅立叶变换红外光谱采用BrukerTensor27spectrotometer型红外光谱仪分析,采用KBr支撑片,在400~4000cm-1范围内记录样品的骨架振动和红外吸收峰。取样品(约2mg)和干燥的溴化钾粉末(约100mg)于玛瑙研钵中,用力将混合物研磨成均匀的极细的粉末,然后在压片机上(107Pa)压成几乎呈透明状的自支撑圆薄片(面积2cm2)。样品薄片的傅里叶红外变换分析在连接计算机的BrukerTensor27spectrotometer红外光谱仪上,在1cm-1的光谱分辨率和128次累计扫描条件下进行分析。催化剂的FT-IR图如图3所示。对于CNT-PVSAIC,傅里叶变换红外光谱在1066cm-1表现出特征峰,对应的是对称N-SO3键的伸缩振动,表明在咪唑环上成功地引入了磺酸基团,强吸收峰1290厘米和1178厘米分别对应的是磺酸基团中不对称的和对称的S-O键的伸缩振动。此外,3430厘米处的宽峰对应磺酸基团中O-H键的伸缩振动。1638厘米和1463厘米处的吸收峰分别对应咪唑环中的C=N键和C=C键的伸缩振动。3110厘米和850厘米处的峰分别对应咪唑环中C–H键的伸缩振动和弯曲振动。在CNT-PVSAPC的傅里叶变换红外光谱中,1054厘米,1162厘米和1235厘米处的三个特征峰分别对应N–SO2键的振动模式,O–SO2键的对称和非对称伸缩振动模式。由此,证实了SO3H基团与吡啶环中N之间N-SO3H键的形成。3417厘米处的宽吸收峰相对应的是SO3H基团中O-H的伸缩振动。C=C键,C=N键的振动峰分别在1506厘米和1652厘米处。另一方面,PVSAPC段的吡啶环中的C-H键伸缩振动和弯曲振动的峰位置分别为3123厘米和850厘米。以上提到的各组峰证实了[C5H5N–SO3H]Cl结构的存在。最后,CNT-PSSA的傅里叶变换红外光谱与文献资料结果吻合。1593厘米和1392厘米处的强特征峰分别表示苯基的伸缩振动和磺酸基团。同样的,以3445厘米为中心的宽带峰对应SO3H基团中–OH官能团的伸缩振动。2918厘米和2816厘米处的特征峰分别对应烷烃中C–H键的非对称和对称的振动吸收。由此,上述的傅里叶变换红外光谱结果证实了在所制得的CNT-P-SO3H复合材料中聚合物P-SO3H的存在。实施例2催化剂的比表面积,孔径分布及孔容的测定在SI-MP-10/PoreMaster33,Quantachrome吸附仪上完成。采用液氮温度77K(-196℃)下的氮气吸脱附测得BET比表面积和孔径分布,样品均于120℃高真空下预处理24h。在0.05<P/P0<0.3范围内采用Brunauer-Emmett-Teller(BET)计算比表面积;采用Barrett-Joyner-Halenda(BJH)方法来计算样品的孔径分布曲线,通过DensityFunctionalTheory(DFT)方法确定孔容。如图4所示。以相对压力为X轴,氮气吸附量为Y轴,再将X轴相对压力粗略地分为低压(0.0-0.1)、中压(0.3-0.8)、高压(0.90-1.0)三段。那么从图4中可以得知,吸附曲线在低压端X轴,则说明三种催化剂材料CNT-PVSIAC,CNT-PVSAPC,CNT-PSSA以及CNT与氮气相互作用力较弱;中压端多为氮气在催化剂骨架支撑体内的积聚,通过它可以判断为介孔分析。BJH方法就是基于这一段得出的孔径数据;高压段推测出所测样品中分子的堆积程度。滞后环的特征对应于特定的孔结构信息。滞后环的产生原因,这是由于毛细管凝聚作用使N2分子在低于常压下冷凝填充了介孔孔道,开始发生毛细凝结时是在孔壁上的环状吸附膜液面上进行,而脱附是从孔口的球形弯月液面开始,因此吸脱附等温线不相重合,往往形成一个滞后环。而图4中催化剂与碳纳米管的滞后环都属于H3型,高压端的吸附量比较大,使得回滞环等温线在较高相对压力区域没有明显饱和吸附平台。表明碳管支撑体的孔道结构不是很规整,认为是由片状粒子一层一层的地不断堆积得到狭长的缝孔。实施例3碳基固体酸催化剂样品的热重分析在TAInstrumentQ600SDT型热重分析仪上进行,升温范围为室温~600℃,升温速率为10K/min,载气流量为50mL/min,试样质量为5~10mg。如图5所示。通过热重-差热分析(TG-DTA,图5)研究了CNT-P-SO3H复合材料的热稳定性。图5显示了CNT,CNT-PVSAIC,CNT-PVSAPC及CNT-PSSA的重量损失随温度的变化曲线。正如预期的,纯碳纳米管直到600℃,其质量损失可忽略不计。但是,CNT-P-SO3H复合材料的高分子降解使其失重明显。CNT-PVSAIC大概在227℃开始明显失重,同理,CNT-PVSAPC大概在214℃其聚合物开始裂解,CNT-PSSA的玻璃化转变温度为309℃。显然,CNT-PSSA比CNT-PVSAIC和CNT-PVSAPC的热稳定性更高。此外,根据它们各自的热重结果,通过比较CNT-P-SO3H复合材料与纯碳纳米管之间的重量损失来估计CNT-P-SO3H复合材料中P-SO3H的负载程度。从TG结果可以看出,CNT-PVSAIC,CNT-PVSAPC,CNT-PSSA中P-SO3H的含量分别是26.6wt%,24.5wt%和8.2wt%。实施例4催化剂的物性表征在BrukerAdvanceD8diffractometer上进行,管电压40KV,管电流40mA,特征射线Cu-Kα辐射光源,小角扫描范围2θ=5~50,扫描步长0.02°,采样时间0.3s。取100℃高真空干燥后的样品(约3mg),采用石英板,样品经磨细,使粉末样品在石英板的填样区的均匀分布并用玻璃板压平实,要求试验面与玻璃表面齐平,压好片后再测试。XRD图如图6所示。在CNT-PVSAIC,CNT-PVSAPC和CNT-PSSA的粉末X射线衍射模式中,只显示了两个特征峰,分别在高强度26゜的2θ值和低强度43゜的2θ值。分别与碳纳米管的特征峰(002)和(100)匹配(图6)。这些结果表明在聚合物官能化碳纳米管的过程中,纯碳纳米管的结晶度结构并没有受到很大的破坏。与CNT相比较,CNT-P-SO3H复合材料的特征衍射峰的强度显著降低,这可归结于CNT-P-SO3H复合材料中的碳纳米管中碳表的微观结构的变化。实施例5拉曼表征使用的仪器是LabRAMHR800-LS55激光拉曼仪。表征在空气气氛中室温下进行,通常采用Ar离子激光源,波长为532nm,激光的光强为30KeV。如图7所示。拉曼光谱用来确定CNT-P-SO3H复合材料聚合过程前后,部分碳纳米管缺损密度的变化情况(如图7)。以纯碳纳米管为例,1342厘米处的峰与无序的石墨结构(D模)中碳原子的振动有关。相对应的是弯曲的石墨片层的缺陷,sp3碳杂化以及其他杂质。而1572厘米处的峰对应的是碳纳米管的石墨结构(G模)。D峰和G峰的强度比(ID/IG),反映了样品中单壁碳纳米管的相对含量和纯度、结构缺陷的相对程度。相较于纯碳纳米管,CNT-P-SO3H复合材料中,经过P-SO3H共价接枝以后碳纳米管的D模与G模的变化可忽略不计。实施例6三种催化剂的形态学分析是在TEM,JEM-2100HR上进行的。样品经研磨后,取少量试样超声分散于无水乙醇中,并用毛细管将高分散的超声液滴在超薄碳网上,待其干燥后直接进行观察。如图8所示。图8显示的是CNT-PVSAIC,CNT-PVSAPC和CNT-PSSA的典型的高分辨率透射电子显微镜图。这些CNT-P-SO3H复合材料的形态近似,它们的HRTEM图像清楚地显示了在碳纳米管上有一层3-5nm的薄高分子涂层,成功证实了高分子官能化碳纳米管(图8)。此外,所有这些CNT-P-SO3H复合材料为准一级结构。相比于纯碳纳米管,能清楚地观察到CNT-P-SO3H复合材料的粗糙表面。尤其,在所得的CNT-P-SO3H复合材料中,P-SO3H专门包裹在碳纳米管的外表面,得到一聚合物层包裹碳纳米管支撑的材料,形成一种名叫“夹克中的纤维”的结构。因此,在制备CNT-P-SO3H前驱体的过程中(CNT-PVI,CNT-PVP和CNT-PSSNa),碳纳米管和相应单体的共价嫁接可能只在碳纳米管的外表面进行。先前报道的数据表明,聚苯胺包裹碳纳米管只发生在碳纳米管外表面,由于在碳纳米管内部限制反应物进入,所以阻碍了苯胺在碳纳米管内的聚合作用。因此,聚合物官能化的碳纳米管外,P-SO3H段包裹在碳纳米管外表面,使得碳纳米管外表面的密集活性位点均匀分布。这种类型的表面结构,利于反应物接触,便于试剂脱离催化剂。此外,聚合物的共价接枝能很好地解释为什么碳纳米管表面比和孔隙率下降得如此多。实施例7表1中可以看出,CNT-P-SO3H复合材料极易催化三乙酸甘油酯与甲醇的酯交换反应,得到显著的转化率。在60℃温度条件下,三乙酸甘油酯的转化率从>99.9%到90.4%(表1,列3-5)。这些转化率远高于与它们相对应的未经过磺酸集团官能化的催化剂前驱体的催化反应结果,例如CNT-PVI和CNT-PVP的催化结果(表1,1-2列)。除此之外,三种CNT-P-SO3H复合材料固体酸催化剂对三丁酸甘油酯与甲醇的酯交换反应也表现出极好的催化性能,使得三丁酸甘油酯的转化率从98.5%到85.4%(表1,9-11列)。正如预期所料,在给定的催化剂负载量和特定的反应条件下,通过比较三乙酸甘油酯和三丁酸甘油酯的甲醇分解反应,短链的三酸甘油酯分子,三乙酸甘油酯的反应速率高于长链三酸甘油酯分子,三丁酸甘油酯的反应速率,此结果与所报道的文献数据吻合。在三乙酸甘油酯和三丁酸甘油酯的甲醇分解反应中,CNT-P-SO3H复合材料的催化活性按CNT-PVSAIC>CNTPVSAPC>CNT-PSSA的顺序减弱(表1,6-8列,12-14列)。除了研究三酸甘油酯与甲醇的酯交换反应,CNT-P-SO3H复合材料催化剂对C18单不饱和油酸与甲醇的酯化反应仍具有很好的催化效果(表1,15-20列)。先前也有报道各种固体酸催化三乙酸甘油酯的甲醇分解反应,在55℃温度下反应3小时,Amberlyst-15和CNT-PANI-S催化三乙酸甘油酯的转化率分别是50.2%和31.4%。在120℃条件下反应4小时,苯磺酸功能化的CNF与Amberlyst-15催化三油酸甘油脂与甲醇的反应,得到油酸甲酯的产率分别为72%和22%。以上结果都证实CNT-P-SO3H复合材料极好的催化活性。最后,从表1中可以看出,不管是三乙酸甘油酯与甲醇的酯交换反应,还是三丁酸甘油酯与甲醇的酯交换反应,抑或是不饱和油酸与甲醇的酯化反应,在相同反应条件下,相比于催化剂CNT-PVSAPC和CNT-PSSA,催化剂CNT-PVSAIC表现出更好的催化活性。CNT-PVSAIC也是本次研究的主催化剂。三酸甘油酯与甲醇的酯交换反应:油酸与甲醇的酯化反应:表1各种CNT-P-SO3H作用于酯交换反应和酯化反应序号催化剂R时间,[h]转化率,[%]1CNT-PVIMe1012.32CNT-PVPMe1015.13CNT-PVSAICMe10>99.94CNT-PVSAPCMe10>99.95CNT-PSSAMe1090.46CNT-PVSAICMe585.87CNT-PVSAPCMe581.38CNT-PSSAMe575.29CNT-PVSAICnPr1098.510CNT-PVSAPCnPr1097.611CNT-PSSAnPr1085.412CNT-PVSAICnPr581.613CNT-PVSAPCnPr578.114CNT-PSSAnPr570.315CNT-PVSAICC17H331093.016CNT-PVSAPCC17H331086.317CNT-PSSAC17H331080.918CNT-PVSAICC17H33577.219CNT-PVSAPCC17H33569.120CNT-PSSAC17H33563.6Reactionconditions:Catal.(20mg),triglycerideoroleicacid(1.0mmol),methanol(1.2mL),60℃,(Entries1–14forScheme1aandEntries15–20forScheme1b).反应条件:催化剂(20mg),三丁酸甘油酯或油酸(1.0mmol),甲醇(1.2mL),60℃,(第1组到第14组为反应1a即酯交换反应,第15组到第20组为反应1b即酯化反应)。图9中(a)CNT-PVSAIC催化三丁酸甘油酯与甲醇的酯交换反应的产物分布;(b)温度对三丁酸甘油酯与甲醇的酯交换反应的影响;(c)三丁酸甘油酯与甲醇的摩尔比对三丁酸甘油酯甲醇分解反应的影响。图10是催化剂循环回收。实施例8图9(a)反应条件:三丁酸甘油酯(302mg,1.0mmol),CNT-PVSAIC(20mg),甲醇(1.2mL),温度60℃。图9(a)为CNT-PVSAIC作为催化剂用于三丁酸甘油酯的甲醇分解的一个典型的酯交换反应。三丁酸甘油酯能很快地转化成丁酸甲酯和甘油二酸酯。128分钟后,甘油二酸酯会立即进一步转换成甘油一酸酯,所以此时甘油二酸酯的产率达到最大。随后,它的产率会随甘油一酸酯的产生而下降。图9(b)反应条件:三丁酸甘油酯(302mg,1.0mmol),CNT-PVSAIC(20mg),甲醇(1.2mL),温度30℃和60℃。从图9(b)可以看出,即便在较低的温度下反应,只要反应时间足够长酯交换反应同样可以达到较高的转化率,温度对酯交换反应的影响并不是特别大,反应温度的提高可以有效缩短酯交换反应的时间,但是当反应温度超过甲醇的沸点之后,体系内甲醇沸腾造成部分甲醇损失,整个体系达到能量平衡,即使继续提高反应温度,反应瓶内的温度几乎不再上升,造成大量热能的损失和浪费,在工业应用中增加反应的成本。所以,反应温度选择在甲醇沸点左右比较适宜,即65℃。反应温度对CNT-PVSAIC催化的酯交换反应的影响表明在低于甲醇沸点的温度范围内,提高反应温度会促进反应进行。温度越高,反应速率越快。图9(c)反应条件:三丁酸甘油酯(302mg,1.0mmol),CNT-PVSAIC(20mg),甲醇(分别为0.2,1.2和2.5mL),温度50℃。影响酯交换反应的另一个重要因素是甲醇与三酸甘油酯的摩尔比,由此进一步研究甲醇与三丁酸甘油酯的摩尔比。虽然甲醇与三丁酸甘油酯反应时的化学计量比为3:1,但是酯交换反应是可逆反应,过量的甲醇能使可逆平衡反应向生成脂肪酸甲酯的方向移动,由此实际生产过程中醇油摩尔比大于化学计量比。图9(c)列出了不同醇油摩尔比对三酸甘油酯转化率的影响。从图中可以看出,在特定的反应温度和定量的CNT-PVSAIC的条件下,测试了三丁酸甘油酯与甲醇摩尔比为(1:60,1:29,1:5)的三种情况。显然,甲醇的摩尔比越高,酯转化率越高,过量的醇会促进酯交换反应进行。实施例9图10反应条件:CNT-PVSAIC(15mg),三丁酸甘油酯(302mg,1.0mmol),甲醇(1.2mL),60℃,4h。作为一种用于酯交换反应的固体催化剂,其与传统均相酯交换反应催化剂相比最大的优点就是可回收再利用,但是负载型固体催化剂中活性组分的流失是导致催化剂失活的一个主要原因。通过三丁酸甘油酯与甲醇的酯交换反应研究CNT-PVSAIC的重复利用性。将回收且处理好的催化剂于相同的反应条件下,在三丁酸甘油酯较低的转化率情况下进行了6次循环回收实验,以此来深入探究催化剂的稳定性。正如图10所示,经过6次循环回收,测得三丁酸甘油酯的转化率从52.4%降到40.9%,表明催化剂CNT-PVSAIC在循环回收过程中失去部分活性。此现象可能是由于高分子层溶胀紧接着碳纳米管上共价连接的PVSAIC链的部分断裂,导致在循环回收过程中,CNT-PVSAIC的一部分催化活性中心(-SO3H)缺失。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1