一种聚肽的快速检测方法与流程
本发明涉及生化分析检测领域,具体涉及一种聚肽的快速检测方法。
背景技术:
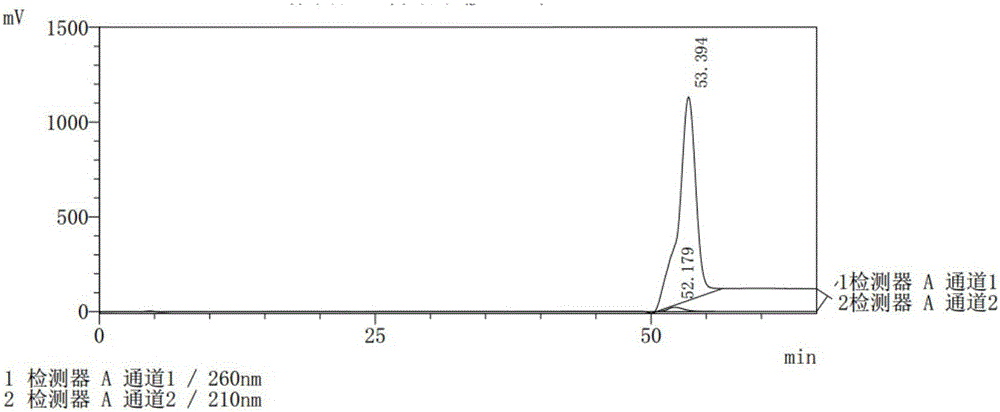
:聚阴离子聚肽的盐溶液(如钠盐、钾盐等)极性强、水溶性好,用传统的C18色谱填料分离分析保留时间短,很难将其与杂蛋白或核酸等杂质分开。电泳检测只能定性,不能定量。高效快速地检测发酵和纯化过程中聚肽含量不仅能提高筛选聚肽高产菌株和纯化的效率,并且对发酵和纯化条件的优化、工艺控制及质量标准的建立有着非常重要的意义。目前常用于测定发酵或纯化过程中聚肽含量的方法通常为水解法,将聚肽水解为对应的单个氨基酸,测定水解前后对应氨基酸含量之差,水解前后样品中氨基酸的含量可通过生物传感分析仪如L-谷氨酸氧化酶电极或HPLC等方法测定(施庆珊,等:一株不需谷氨酸的产聚γ-谷氨酸菌株的筛选与鉴定;孟杰,等:酶法快速检测发酵液中的γ-聚谷氨酸浓度),也有用凝胶色谱测定γ-PGA含量的(YaoJ,etal.RemovalofCr(III),Ni(II)andCu(II)bypoly(γ-glutamicacid)fromBacillussubtilisNX-2[J].)。张庆庆等在《发酵液中γ-聚谷氨酸含量快速测定方法研究》中用十六烷基三甲基溴化铵(CTAB)溶液与γ-PGA反应产生浑浊的特性,通过反应体系的吸光度来反映其浊度,进而通过浊度与γ-PGA浓度的线性关系,测定发酵液中γ-PGA。现有技术中的水解法过程繁琐、水解程度对结果准确性影响较大;凝胶色谱法是根据分子量大小不同保留时间不同来检测的,由于大分子相近分子量的物质较多,凝胶色谱本身的分辨率有限,所以也存在较大的误差;而CTAB法中的CTAB很容易与杂蛋白反应产生浑浊,进而影响检测的准确性。因此,很有必要建立一种聚肽快速、准确的检测方法。技术实现要素:本发明的目的是针对现有技术上的不足,提供了一种聚肽的快速检测方法。为实现以上目的,本发明通过以下技术方案予以实现:一种聚肽的快速检测方法,包括如下步骤:(1)样品溶液的配制:取发酵液或发酵菌体破碎液或纯化中间过程溶液适量,过0.22μm滤膜;(2)样品的测定:采用高效液相离子交换色谱法,以10-30mM的Na2HPO4-NaH2PO4缓冲溶液或K2HPO4-KH2PO4缓冲溶液或Tris-HCl缓冲溶液作为洗脱液A,调pH至7.5-8.5,以洗脱液A为溶剂配制1-3M的NaCl溶液或KCl盐溶液为洗脱液B;所述离子交换色谱条件为:填料QSepharoseTMFF装填于空管柱或使用预装柱;柱温:25℃;进样量:10μL;流速:0.1-2mL/min;洗脱方式:0-10min,100%A等度洗脱;10-25min,洗脱液A:洗脱液B为(85-95):(15-5);25-45min,洗脱液A:洗脱液B为(75-85):(25-15);45-60min,洗脱液A:洗脱液B为(60-75):(25-40);60-80min,100%B等度洗脱,之后通过双波长紫外检测器检测;(3)含量分析。本发明聚肽的快速检测方法中含量分析为根据标准曲线法进行聚肽的含量计算;所述标准曲线为称取聚肽标准品,溶于洗脱液A中,配制成一定浓度的标准溶液,分别稀释成一定浓度梯度的稀释液,过0.22μm滤膜,分别进行检测,以标准溶液的质量浓度(x/mg·mL-1)为横坐标,色谱峰面积为纵坐标(y),绘制标准曲线。本发明所述的离子交换空管柱或预装柱内径为2-20mm,长度为20-100mm,总体积为1-25mL。本发明所述的双波长检测,其中一个检测通道波长设置为205-230nm,用于检测不含苯环的蛋白类物质(包括聚肽),另一个检测通道波长设置为250-280nm,用于检测核酸类和含苯环的蛋白类物质。本发明所述的聚肽可为盐型阴离子聚肽;所述盐型阴离子聚肽包括α-聚谷氨酸、γ-聚谷氨酸、聚天冬氨酸以及由谷氨酸和天冬氨酸所形成的聚肽的盐。进一步地,本发明所述聚肽为α-聚谷氨酸。本发明采用离子交换色谱检测聚肽类物质,并将其应用于HPLC检测平台,不但使聚肽能与其他杂蛋白和大部分核酸等杂质很好地分离,而且采用双波长可以同时检测聚肽和核酸,将核酸的影响去除,样品用量少,只需5-20μL,具有较好的线性范围、重复性和加样回收率,较低的检测限。本发明操作简单,灵敏度高,能快速检测样品中的聚肽。附图说明图1是峰面积-质量浓度曲线;图2是α-聚谷氨酸标准图谱;图3是α-聚谷氨酸发酵菌体破碎液离子交换检测色谱图;图4是α-聚谷氨酸的MALDI-TOF-MS图谱;图5是α-聚谷氨酸初步纯化离子交换检测色谱图。具体实施方式下面结合具体实施例进一步说明本发明的技术解决方案,实施例不能理解为是对技术解决方案的限制。实施例1:标准曲线的绘制1.1色谱检测条件仪器:岛津高效液相色谱仪色谱柱:4.6*50mm,QSepharoseTMFF阴离子交换填料检测波长:210nm,260nm洗脱液A:20mMNa2HPO4-NaH2PO4缓冲溶液,调节pH为8.0洗脱液B:2MNaCl溶液(用20mMNa2HPO4-NaH2PO4缓冲溶液作为溶剂配制)流速:0.3mL/min进样量:10μL柱温:25℃洗脱程序如表1所示:表1洗脱程序1.2线性范围精密称取α-聚谷氨酸标准品10mg于50mL容量瓶中,加水溶解并定容配制成储备液,精密量取不同体积的标准品储备液,配制成浓度梯度为0.0243mg/mL,0.0729mg/mL,0.2187mg/mL,0.6561mg/mL,1.3122mg/mL,1.9683mg/mL的标准品溶液,精密量取标准品溶液10μL,注入高效液相色谱仪,记录色谱图。以标准溶液的质量浓度(x/mg·mL-1)为横坐标,色谱峰面积(y)为纵坐标,绘制标准曲线,得α-聚谷氨酸回归方程为y=(4E+07)x+21643,R=0.9998,说明α-聚谷氨酸在0.0243-1.9683mg·mL-1范围内与其峰面积呈良好的线性关系,如图1所示。1.3重复性试验精密吸取同一供试溶液如线性关系考察中浓度为0.6561mg/mL的溶液10μL,重复进样6次,记录色谱图,峰面积相对标准偏差(RSD)为0.7%,表明重复性良好,具体数值如表2所示。表2重复性试验序号峰面积125091631224958976324781012424764583524897895625198588RSD(%)0.71.4加样回收率试验取已知α-聚谷氨酸含量的发酵菌体破碎液样品6份,分别精密加入标准品储备液0.50mL,摇匀,精密量取10μL,注入高效液相色谱仪,在1.1所述的色谱条件下进行分析,记录色谱图。计算α-聚谷氨酸的回收率,回收率如表3所示。表3α-聚谷氨酸的回收率1.5检出限考察精密量取1.1542mg/mL的α-聚谷氨酸标准品溶液,加水稀释成0.11542mg/mL,0.011542mg/mL,0.005771mg/mL的样品溶液,各精密吸取10μL注入高效液相色谱仪,记录色谱图,其进样量和信噪比如表4所示。表4进样量和信噪比进样量(μg)信噪比(S/N)1.154275.30.01154211.80.057712.7结果表明:当进样量为0.05771μg时,信噪比为2.7,故检测限为0.05771.实施例2:发酵菌体破碎液中α-聚谷氨酸的含量测定取发酵菌体破碎液适量,0.22μm滤膜过滤,精密量取10μL,按实施例1中1.1所述的色谱条件进样分析,重复进样3次,用峰面积按外标法计算α-聚谷氨酸的含量,并在检测过程收集α-聚谷氨酸所对应的峰,富集的样品做MALDI-TOF-MS测试,如图4,显示分子均一性好,纯度高,其含量具体值如表5所示。表5发酵菌体破碎液中α-聚谷氨酸的含量测定实施例3:初步纯化样品中α-聚谷氨酸的含量测定取初步纯化样品适量0.22μm滤膜过滤,精密量取10μL,按实施例1中的色谱条件进样分析,重复进样3次,用峰面积按外标法计算α-聚谷氨酸的含量,如图5所示,其含量具体值如表6所示。表6初步纯化样品中α-聚谷氨酸的含量测定以上所述仅是本发明的优选实施方式,应当指出,在不脱离本发明原理的前提下,还可以做出若干修饰和改进,这些修饰和改进也应视为本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1