电化学还原法制备高量子产率电化学发光金纳米团簇探针的制作方法
本发明涉及一种高量子产率电化学发光金纳米团簇探针材料及其电化学还原制备方法,属于纳米技术领域。
背景技术:
近年来,半导体纳米晶体或量子点作为一种新的电化学发光纳米发光体,由于其稳定性好、抗光漂白能力强、激发光谱较宽等优点已受到广泛关注。半导体纳米晶体或量子点电化学发光结合了半导体、电化学和化学发光等优势成为电致化学发光分析中的新领域而发展迅速。然而毒性强和非分子幂率荧光间歇现象等缺陷给跟踪和成像尤其是生物体内应用等研究带来困难,因此选择灵敏度高、生物相容性好的标记物用于生物分析和医学检测已迫在眉睫。
金纳米团簇作为新型纳米发光体,具有尺寸小、无毒、水溶性好、特殊光电性质等特点,己在临床分析、生物医药、生物传感和催化等领域广泛应用。尽管金纳米团簇的荧光性能及其应用已被广泛研究,但是由于弱的电化学发光强度以及未知的机理,基于金纳米团簇的电致化学发光传感研究还非常少。因此,制备具有强的电化学发光信号和高量子产率的电致化学发光金纳米团簇探针对于构建高性能电致化学发光传感器具有重要意义。
本发明采用简单的电化学还原法制备电化学发光金纳米团簇探针,本发明所述方法工艺简单、反应条件温和、重现性好,所制备的金纳米团簇探针电致化学发光信号强而稳定,且量子产率高。
技术实现要素:
本发明的目的在于提供一种电化学还原法制备高量子产率电化学发光金纳米团簇探针。
为了实现上述目的,本发明采用以下技术方案:
本发明所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是采用恒电位还原法对金纳米团簇材料进行还原得到金纳米团簇探针,还原电位为-0.2 V~-2.0 V,金纳米团簇探针具有良好的电化学发光性能。
所述金纳米团簇材料为功能化修饰金纳米团簇,所述的功能化修饰金纳米团簇采用N-乙酰化-L-半胱氨酸-金纳米团簇,谷胱甘肽-金纳米团簇或牛血清白蛋白-金纳米团簇。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是将金纳米团簇探针修饰在玻碳电极上,并将其作为工作电极,以过硫酸钾为共反应剂,进行电化学发光测试,能产生电化学发光信号。
所述的N-乙酰-L-半胱氨酸-金纳米团簇的合成步骤如下:将浓度为0.1~0.8 mol/L的氢氧化钠与浓度为0.01~0.1 g/L氯金酸溶液加入到浓度为0.02~0.18 mol/L的N-乙酰-L-半胱氨酸溶液中,混匀后置于20~70 ℃恒温水浴恒温反应0.1~3.5小时,待反应结束后透析纯化处理,得到N-乙酰-L-半胱氨酸-金纳米团簇水溶液,冷冻干燥后可得到N-乙酰-L-半胱氨酸-金纳米团簇材料粉末,N-乙酰-L-半胱氨酸-金纳米团簇水溶液进行荧光光谱分析,其最大激发波长和发射波长分别为355 nm和650 nm。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是电化学发光信号由以下方法采集的:将玻碳电极用Al2O3粉末抛光至光滑镜面,再依次放入HNO3水溶液,无水乙醇,去离子水中超声清洗,N2吹干;将金纳米团簇探针修饰在处理好的玻碳电极表面,得到金纳米团簇探针修饰玻碳电极;采用三电极体系进行测试,以金纳米团簇探针修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,缓冲溶液为磷酸盐缓冲液或Tris-HCl缓冲溶液,所用电解质为KCl或KNO3;将上述三电极插入含有过硫酸钾共反应剂的缓冲溶液中,施加一定的电压,工作电极表面产生电化学发光辐射。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是连续电化学扫描24段以上电化学发光信号保持不变。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是电化学发光信号强弱与制备电化学发光金纳米团簇探针所使用的还原电位相关。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是电化学发光信号强弱与电化学发光金纳米团簇探针中Au(0)的比例成线性关系。
所述的电化学还原法制备高量子产率电化学发光金纳米团簇探针,其特征是采用恒电位还原法对金纳米团簇材料进行还原得到,还原电位为-2.0 V,得到的电化学发光金纳米团簇探针的相对电化学发光效率为4.11%。
本发明所述的高量子产率电化学发光金纳米团簇探针的制备方法,其特征是以金纳米团簇材料修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,在磷酸盐缓冲溶液中施加-0.2 V~-2.0 V电压对金纳米团簇材料进行恒电位还原得到。
具体地说,本发明采用以下技术方案:
(一)N-乙酰-L-半胱氨酸-金纳米团簇的制备
N-乙酰-L-半胱氨酸-金纳米团簇合成步骤如下:将浓度为0.1~0.8 mol/L的氢氧化钠与浓度为0.01~0.1 g/L氯金酸溶液加入到浓度为0.02~0.18 mol/L的N-乙酰-L-半胱氨酸溶液中,混匀后置于20~70℃恒温水浴恒温反应0~3.5小时。待反应结束后透析纯化处理,得到N-乙酰-L-半胱氨酸-金纳米团簇水溶液,冷冻干燥后可得到N-乙酰-L-半胱氨酸-金纳米团簇材料粉末。
(二)金纳米团簇修饰电极的制备
将玻碳电极用1.0 μm、0.3 μm和0.05 μm的Al2O3粉末依次抛光,打磨,至光滑镜面,再依次放入HNO3溶液(1:1),无水乙醇,去离子水中超声清洗3分钟,N2吹干。取5 μL N-乙酰-L-半胱氨酸-金纳米团簇水溶液滴加在处理好的玻碳电极表面,室温干燥,即得N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极。
(三)电化学还原处理金纳米团簇
采用三电极体系进行还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 mol/L pH 7.4的磷酸盐缓冲溶液中,施加不同负电位电压(-0.2 V ~ -2 V范围内),进行恒电位还原处理,得到电化学发光金纳米团簇探针。
(四)金纳米团簇探针电致化学发光信号的产生和检测
采用三电极体系进行测试,以金纳米团簇或金纳米团簇探针修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入含有共反应剂的缓冲溶液中,采用阶跃脉冲法,初始电位为0 V,脉冲时间为10 s,终止电位为-2 V,脉冲时间为1 s。光电倍增管高压设置为600 V ~ 800 V,检测工作电极表面产生的电致化学发光信号,采用电化学还原法制备的金纳米团簇探针其电致化学发光信号显著增强。
本发明的优点是:
(1)本发明以功能化修饰金纳米团簇为前驱体,采用电化学方法进行还原制备高性能电化学发光金纳米团簇探针,该方法具有制备方法绿色环保、操作简便、重现性好等优点。
(2)本发明所得到的电化学发光金纳米团簇探针发光强度大,电化学发光量子产率高,生物相容性好,在电致化学发光传感器件和生物医学领域具有良好的应用前景。
附图说明
图1为N-乙酰-L-半胱氨酸-金纳米团簇的荧光光谱图。
图2为N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极的电致化学发光-时间曲线图。
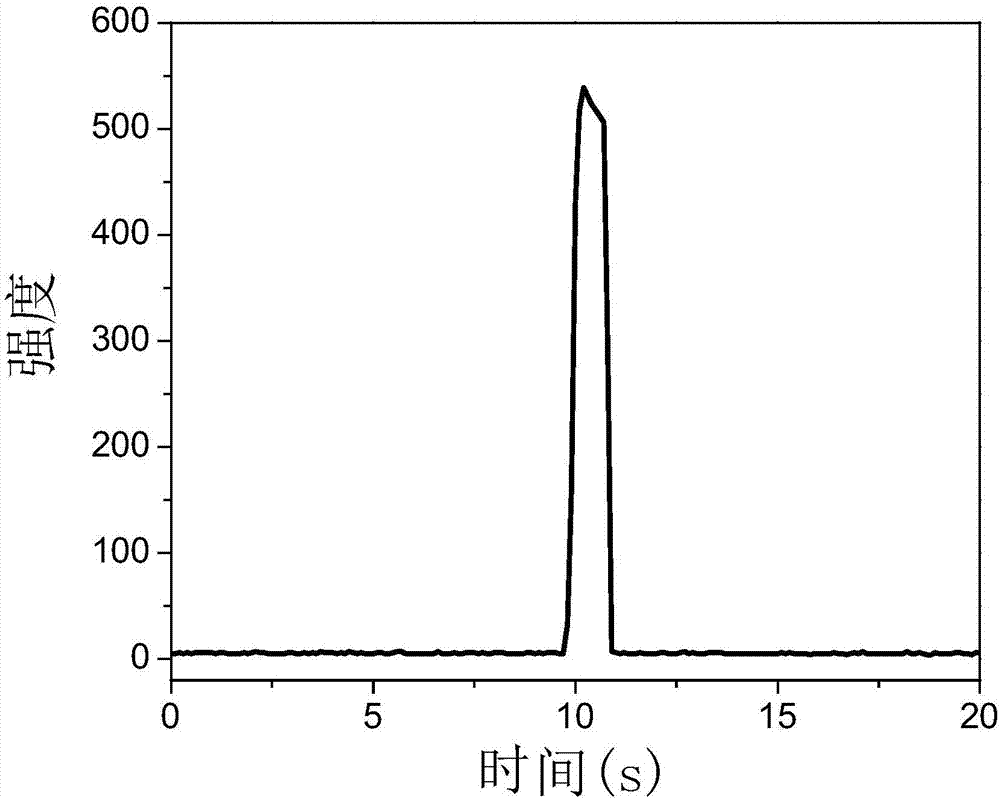
图3为电化学还原法制备的金纳米团簇探针修饰玻碳电极的电致化学发光-时间曲线图。
图4为电化学还原法制备的金纳米团簇探针修饰玻碳电极连续扫描24段所得的电化学发光强度图。
图5为金纳米团簇探针的制备电位对金纳米团簇探针中Au(0)比例的影响图。
图6为金纳米团簇探针的制备电位对金纳米团簇探针电化学发光强度的影响图。
图7 为金纳米团簇探针电化学发光强度与金纳米团簇探针中Au(0)比例的线性关系图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步阐述,本发明并不限于此。
实施例1
往4 mL浓度为0.08 mol/L的N-乙酰-L-半胱氨酸溶液中加入0.6 mL浓度为0.5 mol/L的氢氧化钠与0.4 mL浓度为20 mg/mL氯金酸溶液,混匀后置于37 ℃恒温水槽中孵育3小时。反应结束后的反应液用截留分子为3500的透析袋进行透析纯化处理,得到N-乙酰-L-半胱氨酸-金纳米团簇水溶液,冷冻干燥后得到N-乙酰-L-半胱氨酸-金纳米团簇粉末。取上述N-乙酰-L-半胱氨酸-金纳米团簇水溶液进行荧光光谱分析,可得最大激发波长和发射波长分别为355 nm和650 nm(见图1)。
实施例2
将直径3 mm的玻碳电极依次用1.0 μm、0.3 μm和0.05 μm的Al2O3粉末抛光至光滑镜面,再依次放入HNO3溶液(浓硝酸与水体积比为1:1),无水乙醇,去离子水中超声清洗3分钟,N2吹干。取5 μL N-乙酰-L-半胱氨酸-金纳米团簇溶液滴加在处理好的玻碳电极表面,室温干燥,得N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极。将上述电极插入含有0.1 mol/L过硫酸钾和0.1 mol/L KCl的0.1 mol/L pH 7.4的磷酸盐缓冲溶液中。采用阶跃脉冲法,初始电位为0 V,脉冲时间为10 s,终止电位为-2 V,脉冲时间为1 s。光电倍增管高压设置为700 V,检测工作电极表面产生的电致化学发光信号,得到较弱的电化学发光信号(见图2)。
实施例3
采用三电极体系进行电化学还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 mol/L pH 7.4的磷酸盐缓冲溶液中,进行恒电位还原,还原电位为-2.0 V,还原时间为5分钟,得到还原金纳米团簇探针修饰玻碳电极。将制备的还原金纳米团簇探针修饰玻碳电极插入含有0.1 mol/L过硫酸钾和0.1 mol/L KCl的0.1 mol/L pH 7.4的磷酸盐缓冲溶液中。采用阶跃脉冲法,初始电位为0 V,脉冲时间为10 s,终止电位为-2 V,脉冲时间为1 s。光电倍增管高压设置为700 V,检测工作电极表面产生的电致化学发光信号(见图3),其信号约为未还原处理的N-乙酰-L-半胱氨酸-金纳米团簇的30倍。
实施例4
采用三电极体系进行电化学还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 mol/L pH 7.4的磷酸盐缓冲溶液中,进行恒电位还原,还原电位为-2.0 V,还原时间为5分钟,得到金纳米团簇探针修饰玻碳电极。将制备的金纳米团簇探针修饰玻碳电极插入含有0.1 mol/L过硫酸钾和0.1 mol/L KCl的0.1 M pH 7.4的磷酸盐缓冲溶液中。采用阶跃脉冲法,初始电位为0 V,脉冲时间为10 s,终止电位为-2 V,脉冲时间为1 s。光电倍增管高压设置为700 V,连续扫描24段,记录电致化学发光信号(见图4),其电致化学发光信号保持不变。
实施例5
采用三电极体系进行电化学还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 mol/L pH 7.4磷酸盐缓冲溶液中,进行恒电位还原,还原电位分别为-0.2V、-0.5 V、-0.8 V、-1.0 V、-1.2 V、-1.5 V、-1.7 V、-1.8 V和-2.0 V,得到不同还原程度的金纳米团簇探针修饰玻碳电极。再对上述不同还原电位条件下还原处理的金纳米团簇探针修饰在可拆玻碳电极表面进行X射线光电子能谱检测,得到Au(0)含量。如图5所示,Au(0)含量随着还原电位降低而增大,还原电位为-1.5 V时,Au(0)含量接近100%,表明+1价Au全部被还原为零价Au。
实施例6
采用三电极体系进行电化学还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 M pH 7.4磷酸盐缓冲溶液中,进行恒电位还原,还原电位分别为-0.2 V、-0.5 V、-0.8 V、-1.0 V、-1.2 V、-1.5 V、-1.7 V、-1.8 V和-2.0 V,得到不同还原程度的金纳米团簇探针修饰玻碳电极。将制备的还原金纳米团簇探针修饰玻碳电极插入含有0.1 mol/L过硫酸钾和0.1 mol/L KCl的0.1 mol/L pH 7.4 磷酸盐缓冲溶液中。采用阶跃脉冲法,初始电位为0 V,脉冲时间为10 s,终止电位为-2 V,脉冲时间为1 s。光电倍增管高压设置为700 V,分别记录不同还原电位处理的金纳米团簇修饰电极的电致化学发光信号。如图6所示,上述金钠米团簇探针修饰电极电致化学发光信号强度与还原电位成正比,还原电位为-1.5 V时达到最大,并趋于稳定。同时,对上述不同还原电位条件下还原处理得到的金纳米团簇探针进行X射线光电子能谱(XPS)测试,得到相应Au(0)的含量。如图7所示,电化学发光强度与Au(0)含量成良好的线性关系。
实施例7
采用三电极体系进行电化学还原,以N-乙酰-L-半胱氨酸-金纳米团簇修饰玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将上述电极插入0.1 M pH 7.4磷酸盐缓冲溶液中,进行恒电位还原,还原电位为-2.0 V,还原时间为5分钟,得到还原金纳米团簇探针修饰玻碳电极。将制备的金纳米团簇探针修饰玻碳电极插入含有0.1 mol/L过硫酸钾和0.1 mol/L KCl的0.1 mol/L pH 7.4 磷酸盐缓冲溶液中。采用循环伏安法,施加0 V ~ -2.0 V的线性扫描电压,扫描速度为0.2 V/s,光电倍增管高压设置为700 V,检测工作电极表面产生的电致化学发光信号(I),其产生的相应电量为Qf。另外,将直径3 mm的玻碳电极用1.0 μm、0.3 μm和0.05 μm的Al2O3粉末依次抛光,打磨,至光滑镜面,再依次放入HNO3溶液(浓硝酸与水体积比为1:1),无水乙醇,去离子水中超声清洗3分钟,N2吹干。采用三电极体系,以裸玻碳电极为工作电极,铂丝电极为对电极,Ag/AgCl为参比电极,将裸玻碳电极插入含有1.0 mmol/L [Ru(bpy)3]2+和0.1 mol/L四丁基高氯酸铵的乙腈溶液中,施加-1.0 V ~ -1.8 V的线性扫描电压,扫描速度为0.2 V/s,光电倍增管高压设置为700 V,检测工作电极表面产生的电致化学发光信号(I°)其产生的相应电量为Q°f。由公式ΦECL=Φ°ECL(IQ°f/ I°Qf)计算得到电化学还原法制备的金纳米团簇探针的电化学发光效率ΦECL为4.11%。
以上所述仅为本发明的典型实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改,等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!