一种湖泊沉积物中单酯磷组分的提取及磷核磁共振谱图分析方法与流程
本发明属于环境化学分析测试方法技术领域,具体涉及一种湖泊沉积物中单酯磷组分的提取及磷核磁共振谱图分析方法。
背景技术:
磷是水生生态系统关键的限制性营养元素之一,过量的磷导致藻类大量生长繁殖,引起水质恶化并危害生态系统安全。近年来,包括生活污水、工农业生产、土壤径流等过程携带的外源磷输入得到了一定的控制,湖泊内源磷的释放成为引起湖泊水质进一步恶化的关键因素。沉积物释放是湖泊内源磷污染重要的来源之一,而有机磷是湖泊沉积物中磷的重要组成部分。已有研究表明,有机磷(po)在水生生态系统的磷循环中扮演重要角色,一些po组分可通过酶水解等过程转化为生物可利用性活性磷酸盐,进而成为富营养化湖泊蓝藻水华持续暴发的重要磷源。
当前,沉积物中有机磷分析方法包括磷31核磁共振技术(31p-nmr)、酶水解技术、基于同步辐射光源x射线吸收精细结构分析技术、液相色谱或离子色谱与电感耦合等离子发射光谱连用(hplc-icp-aes)及液相色谱或离子色谱与电子喷雾离子化质谱联用技术(hplc-esi-ms/ms)等。worsfold等人在2008年发表的文章《characterisationandquantificationoforganicphosphorusandorganicnitrogencomponentsinaquaticsystems:areview》(analyticachimicaacta.624(1):37-58(2008))中详细介绍了各种分析检测技术。近年来,31p-nmr作为定性定量分析手段在湖泊沉积物样品的磷组分研究中得到广泛运用,获得了一些沉积物磷组分的基本信息,如正磷酸盐、磷酸单酯、磷酸二酯、焦磷酸盐、多聚磷酸盐、磷脂和膦酸盐等在沉积物中的含量及部分结构组成信息。
但是,湖泊沉积物提取液31p-nmr谱图单酯磷区域中含有大量不同结构的有机磷化合物,如肌醇六磷酸盐同分异构体包括肌-肌醇六磷酸盐、鲨-肌醇六磷酸盐和新-肌醇六磷酸盐,葡萄糖磷酸盐以及腺苷单磷酸盐等。这些单酯磷在液相31p-nmr谱图中的化学位移基本介于3.0ppm至6.5ppm小范围之内(以正磷酸盐化学位移定为6.0ppm为基础)。一方面,沉积物中有机磷含量较低;另一方面,这些有机磷峰受提取液中ph及顺磁性金属离子(如fe3+等)影响,各峰之间相互重叠,峰展宽。这导致了沉积物提取液31p-nmr谱图中单酯磷区域的分辨率不高,甚至对谱图中单酯磷化合物的错误解析。尤其是我国幅员辽阔,不同湖泊沉积物基本性质差异很大,不同沉积物样品提取液中背景金属离子等含量差异很大,导致磷化学位移存在差异,尤其是31p-nmr谱图中单酯磷具体组分的判断造成了很大的困难或误判。
为了消除磁性离子对31p核磁共振分析精确度的干扰,现有技术中已有相关报道,如中国专利《用31p核磁共振分析环境样品时消除磁性离子干扰的方法》即公开了一种采用8-羟基喹啉沉淀去除含磷提取液中大量的铁、锰等磁性离子,从而大幅度降低31p-nmr波谱峰宽,显著提高磷组分的鉴别及定量分析的方法。该方法消除磁性离子干扰的效果显著,且在处理过程中不造成磷组分的损失,操作简便、成本低。
又如中国专利《一种钙质沉积物有机磷的提取及组成分析方法》即公开了一种先采用偏酸性的乙二胺四乙酸溶液对沉积物进行冲洗,络合去除部分铁锰等顺磁性金属离子,再结合传统方法使用naoh+edta将沉积物中的有机磷提取出来,通过31p-nmr进一步分析提取液中有机磷的组成。该方法适用于金属元素ca/(fe+al)>0.7的钙质沉积物。
虽然上述分析方法可一定程度上消除顺磁性金属离子对总有机磷31p核磁共振分析的干扰,但并未公开采用31p核磁共振分析单酯磷的具体方法。而且,一方面由于湖泊沉积物在采用碱液(naoh、edta或其组合)连续长时间提取后获得,而这个长时间提取过程会造成一些不稳定单酯磷的降解;另一方面,一些不稳定的二酯磷也容易大量降解为单酯磷,所以,现在提取和前处理技术对于结合31p-nmr精确分析沉积物中单酯磷具有局限性。
更为关键的是,现有技术中,对湖泊沉积物提取液31p-nmr谱图中单酯磷的解析主要采用以下方法:(1)以正磷酸盐化学位移为参考,调节单酯磷化学位移,参考已知文献报道各单酯磷化学位移,对各单酯磷进行定性和定量分析;(2)提取液中直接加入磷标准物质,如亚甲基二磷酸盐(mdpa,methylenediphosphonicacidsodium),以该磷标准物质化学位移为参考,调节单酯磷化学位移并参考文献报道,对各单酯磷进行定性和定量分析;(3)与沉积物提取同步,设定一个加入有机磷标准样品的背景溶液,同步处理后,分析其中有机磷标准化合物化学位移,以此为参考并结合文献报道,判断沉积物提取液中各单酯磷组分。然而,实践证明,这些分析方法存在以下问题:(1)对比发现,以正磷酸盐化学位移为参考,不同文献报道31p-nmr谱图磷化合物化学位移均存在差异。尤其是单酯磷,由于化学位移集中且相互靠近,以正磷酸盐化学位移为参考,参照不同文献报道的化学位移会得出不同的分析结果;(2)提取液中直接加入磷标准物质,为不影响单酯磷的判别,往往采用远离单酯磷化学位移区域的标准物质,如mdpa,其化学位移为17-18ppm之间,远离单酯磷区域,以此为标准,参照文献报道,也无法准确判断31p-nmr谱图中相应化学位移对应的单酯磷化合物;(3)沉积物中提取液的离子浓度等均会影响磷化合物化学位移,与沉积物提取同步,设定一个加入有机磷标准样品的背景溶液获得的模拟标准样品,其离子浓度与有机质等均与沉积物提取液相差很大,实践证明,二者的化学位移差异很大,尤其是单酯磷区域。
另外,湖泊类型多种多样,不同区域湖泊地质背景差异很大,沉积物基本性质如矿物组成等差异大,导致沉积物有机磷提取液中离子强度、有机质含量等背景值差异大,这直接导致不同沉积物提取液中31p-nmr谱图化学位移差异。因此,以上传统的分析方法中仍没有可靠的方法得到湖泊沉积物提取液31p-nmr谱图中单酯磷组分的准确的分析结果。
技术实现要素:
本发明的目的是提供一种湖泊沉积物中单酯磷组分的提取及磷核磁共振谱图的分析方法,一方面,该方法可以消除顺磁性金属离子对湖泊沉积物中单酯磷组分的干扰,还能最大程度地避免由于有机磷自身的不稳定性引起的降解;另一方面,本申请提供的分析方法避免了单酯磷峰相互重叠及其不稳定性引起的分析结果误差。
为了实现上述目的,本发明提供了一种湖泊沉积物中单酯磷组分的提取及磷核磁共振谱图分析方法,其包括如下步骤:
步骤一,用na2s溶液预处理湖泊沉积物提取浓缩液,得待测液1;
步骤二,对待测液1进行31p-nmr谱图分析;
其中,步骤二的具体操作如下:
(1)测得待测液1的31p-nmr谱图a;
(2)配制50mmol/l的单酯磷标准化合物、二酯磷标准化合物和膦酸盐标准化合物溶液,将其加入步骤1中的待测液1后测得31p-nmr谱图b;
(3)调节31p-nmr谱图a和b的正磷酸盐位移至6.00ppm,比对谱图a和b,确定沉积物中相同物质的峰2-3个,参照31p-nmr谱图b,调节31p-nmr谱图a的化学位移;
(4)基于谱图b中的单酯磷标准化合物化学位移,确定样品谱图a中单酯磷组分;
(5)利用单酯磷的峰面积占总面积的比例及提取液中总磷的含量,计算各单酯磷的含量。
优选地,步骤一中所述的na2s溶液的浓度为0.5-2mol/l;进一步优选为0.8-1.2mol/l;更进一步优选为1mol/l。
其中,所述na2s应相对过量,一方面可沉淀除去溶液中的fe3+、mn2+等顺磁性离子,另一方面可使可能剩余的fe3+还原为fe2+,使沉积物提取浓缩液维持还原环境,还可以除去沉积物中的al3+、ca2+、mg2+,降低溶液盐度,可更好地消除金属离子对31p-nmr分析样品时的干扰。与现有技术中的其它沉淀剂,如8-羟基喹啉相比,不仅省去了加入大量酸中和碱的过程,简化了操作;而且,避免了强酸性物质(8-羟基喹啉溶液由12mol/l浓hcl配制而成)的引入导致的不稳定单酯磷和二酯磷的降解。
进一步优选地,步骤一中待测液1的制备方法具体如下:
向湖泊沉积物提取浓缩液中加入na2s的d2o溶液进行金属离子的沉淀及还原处理,处理后的浓缩液于4℃条件下,静置沉淀15-20h后,2000-10000g条件下,离心分离20-30min,得上清液,即为待测液1。
优选地,所述湖泊沉积物提取浓缩液是采用以下方法提取并浓缩得到:
1)采用naoh与edta的混合溶液多次、短时间和长时间结合连续提取湖泊沉积物样品后合并提取液,所述多次即提取次数不少于3次,短时间即提取时间大于0小于等于4h,长时间即提取时间大于4小于等于16h;
2)测定提取液中的总磷,将提取液浓缩、干燥后加d2o溶解,得湖泊沉积物提取浓缩液。
进一步优选地,步骤1)中naoh与edta的混合溶液的浓度为naoh0.2-0.5mol/l,edta30-55mmol/l;更进一步优选为naoh0.2-0.4mol/l,edta40-55mmol/l,如naoh0.25mol/l,edta50mmol/l。
进一步优选地,步骤1)中使用naoh与edta的混合溶液提取湖泊沉积物样品的次数为3-5次,更进一步优选为3次;每次提取时间逐渐增加,如提取时间依次分别为0.5-2h,2-5h,10-16h;如依次分别为2h,4h,16h。采用多次、短时间和长时间结合、时间逐渐增加的方法连续提取湖泊沉积物样品可以避免因单酯磷及多酯磷的降解引起的31p核磁共振谱图中单酯磷组分分析结果误差。
提取次数小于3次时,要充分提取出样品中的磷,需延长每次提取时间,但由于提取时间过长,提取过程中部分单酯磷、多酯磷降解,导致磷核磁共振谱图组分分析得到的结果误差增大;
提取次数太多,如超过5次时,提取液体积大,浓缩后盐都太高,提取得到的有机磷和单酯磷的再溶解以及后续31p-nmr分析均受到影响,最后的分析结果误差也会显著增加。
进一步优选地,提取时的温度为4-25℃。
进一步优选地,步骤2)中测定提取液中的总磷是采用磷钼蓝分光光度法或者采用电感耦合等离子体发射光谱仪(icp-oes)分析测定。
优选地,步骤(2)中单酯磷标准化合物至少包括肌醇六磷酸盐myo-ihp、葡萄糖6磷酸盐g6p、葡萄糖1磷酸盐g1p、α-甘油磷酸盐α-gly、β-甘油磷酸盐β-gly、单磷酸腺苷amp、胆碱磷酸pcho;二酯磷化合物至少包括dna-p;
膦酸盐标准化合物至少包括亚甲基二磷酸盐mdpa。
优选地,步骤(1)中待测液1的核磁共振仪的条件设置为:温度为25℃,脉冲为45℃,脉冲持续时间为5.3μs,采集时间为0.6s,驰豫时间设置为5s,扫描次数为12000~15000次。
优选地,步骤(3)的操作步骤为:首先,调节31p-nmr谱图a和b的正磷酸盐位移至6.00ppm;然后,调节谱图a和b中化学位移范围为6.5ppm至1.5ppm的信号峰高度至可对谱图a和b中单酯磷区域峰信号进行精细对比;最后,确定沉积物中相同物质的峰2-3个,对比相同物质的峰位移,参照谱图b,调节谱图a中该峰化学位移至一致。
对于步骤(4),当样品中出现标准样品以外的谱峰时,以标准样品化学位移为参考,通过参考已公开文献和相对单酯磷标准化合物标准单酯磷化学位移,分析其它单酯磷的组分,其中单酯磷组分包括:肌醇磷酸盐异构体myo-ihp(inositolhexakisphosphate)、chiro-ihp、neo-ihp和scyllo-ihp;chiro-ihp包括chiro-ihp(2e/4a)和chiro-ihp(4e/2a);d-肌醇1,4,5-三磷酸盐d-myo-itp(d-myo-inositol-1,4,5-triphosphate);α-甘油磷酸盐α-gly(glycerophosphate,β-甘油磷酸盐β-gly(glycerophosphate;葡萄糖磷酸盐g6p(glucose-6-phosphate)、g1p(glucose-1-phosphate);单核苷酸nuc(mononucleotides);磷酸胆碱pcho(cholinephosphate)。
与现有技术相比,本申请提供的湖泊沉积物中单酯磷组分提取及其磷核磁共振谱图分析方法具有如下优点:
(1)本申请在提取过程中加入过量的na2s溶液,可在提取液碱性条件下消除顺磁性金属离子对31p-nmr分析样品时的干扰的同时避免对不稳定性单酯磷和二酯磷的影响,保证了单酯磷组分分析的精确性。
(2)由实施例内容可以得知,本申请采取加标准化合物溶液对比的分析方法,可精确分析得出11种单酯磷组分及其对应含量,而采用上述传统方法至多只能分析得出5-8种单酯组分,甚至会因对化学位移判断错误而获得错误的解析结果。另外,本申请得到的单酯磷组分总含量比传统分析方法高出3%-10%。
(3)本发明的优选方案提取浓缩液的方法可优先提取出稳定性较差的有机磷,尤其是单酯磷,磷提取率明显提高。其中,对沉积物总磷的提取率是传统的一次长时间提取方法(只提取一次,提取时间16h以上)提取率的1-1.5倍;沉积物单酯磷的提取率是传统的一次长时间提取方法提取率的1-2倍,对于湖泊沉积物而言,其提取效果明显优于传统提取工艺。
附图说明
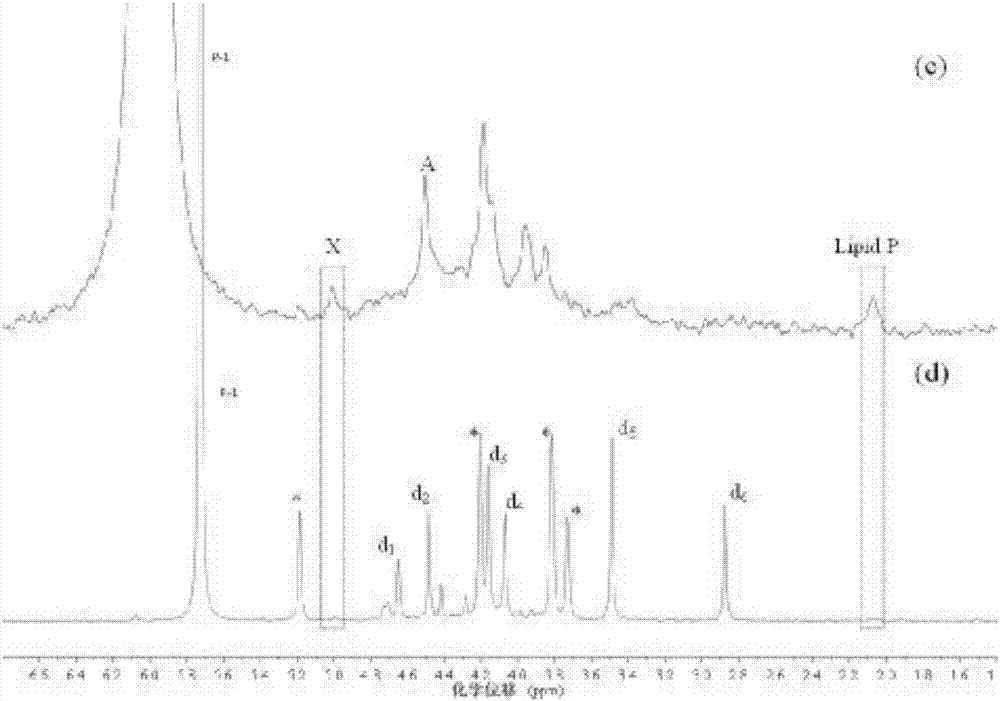
图1实施例1中的dc-1沉积物样品naoh-edta提取液以及加入磷标准化合物31p-nmr谱图单酯磷区域对比
图2实施例1中的dc-1沉积物样品naoh-edta提取液以及加入磷标准化合物31p-nmr谱图调节后单酯磷区域对比
注:①图-1、2中加入的单酯磷标准化合物磷标准物质包括肌醇六磷酸盐(myo-ihp);d1,葡萄糖6磷酸盐(g6p);d2,α-甘油磷酸盐(α-gly);d3,β-甘油磷酸盐(β-gly);d4,5’单磷酸腺苷(amp);d5,胆碱磷酸(pcho);d6,葡萄糖1磷酸盐(g1p);
②图-1、2中(a)和(c)为沉积物naoh-edta提取液31p-nmr谱图;(b)和(d)为对应提取液加入单酯磷标准化合物后31p-nmr谱图。
③图1中31p-nmr谱图对比以正磷酸盐为参考,将其位移调节为6.00ppm;图2中31p-nmr谱图以加入单酯磷标准化合物后的谱图为参照,调节沉积物naoh-edta提取液31p-nmr谱图。
具体实施方式
下面以我国滇池湖泊沉积物样品提取液的磷核磁图谱中单酯磷组分分析为例,对本方法作进一步说明。这些实施例仅是出于解释说明的目的,而不限制本发明的范围和实质。
本发明的所需试剂:
1)碱性提取剂:称取10gnaoh和18.6gedta(na盐)溶解到1.0l去离子水,配置成0.25mnaoh+50mmedta混合液。
2)na2s溶液:称取480mgna2s·9h2o,加2mld2o溶解,配制成1m的na2s/d2o溶液。
3)k2s2o8溶液:将5g过硫酸钾(k2s2o8)溶解于去离子水,并稀释至100ml,配制成50g/l的溶液。其中,采用k2s2o8溶液消解的方法按照gb11893-89中规定的方法执行。
4)磷标准化合物溶液:称取磷标准化合物(包括亚甲基二磷酸盐(mdpa)、肌-肌醇六磷酸盐(myo-ihp)、葡萄糖6磷酸盐(g6p)、葡萄糖1磷酸盐(g1p)、α-甘油磷酸盐(α-gly)、β-甘油磷酸盐(β-gly)、单磷酸腺苷(amp)胆碱磷酸(pcho)和二酯磷(dna-p)),加d2o溶解并将溶液定容后配制成50mm(以磷计)的磷标准化合物溶液。
所需设备:
干冰或冰袋、冷冻干燥机、往复式振荡机、离心机、电感耦合等离子体发射光谱仪(icp-oes)、安捷伦dd2500mhz或600mhz核磁共振仪(仪器配备5mmonenmr探头)。
具体实施方式
实施例1
一种湖泊沉积物中单酯磷组分提取及其磷核磁共振谱图分析方法,其包括如下步骤:
1、采集我国滇池表层沉积物(0-10cm)样品,具体见表1,冷冻干燥后研磨粉碎过100目筛,装入封口袋中,于0至-20℃低温冷冻保存备用;
表1实施例1中湖泊表层沉积物总磷含量与金属元素(al、ca、fe、mg和mn)含量特征
2、称取1.00±0.01g沉积物样品于50ml离心管中,加入8ml0.25m-50mmedta溶液,在室温下(25℃)连续提取三次,每次提取时间分别依次为2h、4h和16h,将三次所得提取液于4℃,10000g条件下离心分离30min后收集、合并上清液,得提取液;
3、采用k2s2o8溶液消解后,分析测定提取液中总磷(tp)含量,或者直接采用电感耦合等离子体发射光谱仪(icp-oes)测定提取液中tp含量;
4、然后使用冷冻干燥机将提取液样品冷冻干燥后得固体粉末,加入2mld2o使样品充分溶解,得沉积物naoh-edta提取浓缩液样品;
5、向步骤4中的提取浓缩液样品中加入0.2ml1mna2s/d2o溶液,然后于4℃条件下,静置沉淀17-20h后,10000g条件下,离心分离30min后得上清液即待测液1;
6、采用安捷伦dd2500mhz或600mhz核磁共振仪获得待测液1的31p-nmr图谱(图1),其中,核磁共振仪的参数设置如下:温度控制在25℃,脉冲为45℃(持续时间为5.3μs),采集时间为0.6s,驰豫时间设置为5s,扫描次数为12000~15000次;
7、配置浓度为50mm(以磷计)磷标准化合物溶液(直接溶解于d2o),具体磷标准化合物包括亚甲基二磷酸盐(mdpa)、肌醇六磷酸盐(myo-ihp)、葡萄糖6磷酸盐(g6p)、葡萄糖1磷酸盐(g1p)、α-甘油磷酸盐(α-gly)、β-甘油磷酸盐(β-gly)、单磷酸腺苷(amp)、胆碱磷酸(pcho)、以及二酯磷dna-p;将所配制的各磷标准化合物溶液分别移取20μl加入步骤1中的待测液1后测得31p-nmr谱图b;
8、采用mestrenova(mnova)软件(9.0.1版本,mestrelabresearchsl)处理31p-nmr谱图,谱图的展宽设置为2hz;调节31p-nmr谱图a和b的正磷酸盐位移至6.00ppm,初步比对谱图a和b中单酯磷区域;然后精细对比谱图a和b中单酯磷区域峰信号,确定沉积物中相同物质的峰2-3个;
9、基于谱图b中标准的单酯有机磷化合物化学位移,调整谱图a中的的单酯磷标准化合物化学位移,确定谱图a中的单酯磷组分;
10、利用各单酯磷的峰面积比例及提取液中总磷的含量,计算各单酯磷的含量。测定结果如下表2。
表2实施例1湖泊沉积物naoh-edta提取及31p-nmr表征获得单酯磷组分含量(mgkg-1)及其相对比例(%)
注:括号中值为各磷形态占naoh-edta提取液tp的百分比例。nuc:(mononucleotides)单核苷酸。
经测定,步骤5得到的待测1中各金属离子去除率分别为:mn2+93%;fe3+98%;al3+96%;ca2+93%;mg2+91%。
对比例1
一种湖泊沉积物中单酯磷组分提取及其磷核磁共振谱图分析方法,与实施例1相比,其区别仅在于:
步骤2、称取1.00±0.01g沉积物样品于50ml离心管中,一次性加入24ml0.25m-50mmedta溶液,在室温下(25℃)提取22h,将提取液于4℃,10000g条件下离心分离30min后收集上清液,得提取液。
经测定,该实施例提取得到的各单酯磷的含量测定结果如下表3。
表3对比例1湖泊沉积物naoh-edta提取及31p-nmr表征获得单酯磷组分含量(mgkg-1)及其相对比例(%)
注:“-”表示未检测到该成分。
对比例2
一种湖泊沉积物中单酯磷组分提取及其磷核磁共振谱图分析方法,与实施例1相比,其区别仅在于:
省略步骤7;
步骤8、采用mestrenova(mnova)软件(9.0.1版本,mestrelabresearchsl)处理31p-nmr谱图,谱图的展宽设置为2hz;调节31p-nmr谱图a的正磷酸盐位移至6.00ppm;
步骤9、基于谱图a中正磷酸盐的化学位移6.00ppm,确定单酯磷化学位移,参考文献报道《improvedpeakidentificationin31p-nmrspectraofenvironmentalsampleswithastandardizedmethodandpeaklibrary》(geoderma.257-258:102-114(2015))中公开的的31p-nmr谱图中单酯磷的化学位移,以正磷酸盐化学位移6.00ppm为相对位移,确定单酯磷组分。
经测定,该实施例提取得到的各单酯磷的含量测定结果如下表4。
表4对比例2与实施例1湖泊沉积物naoh-edta提取及31p-nmr表征单酯磷组分识别及其含量(mgkg-1)及其相对比例(%)比较结果
注:“-”表明未检测到该成分。
由表4对比数据,结合上述实施例1中数据可以看出:单酯磷区域相同的峰,由于位移判断方式的不同,导致不同的单酯磷组分、含量存在较大差异。实施例1分析得到的单酯磷与标准化合物的化学位移一致,结构及含量的测定结果可靠;而对比例2得到的结果与标准物质相差甚远,导致对单酯磷化合物的结构及含量分析完全错误。例如,对比例2中分析得到的新单酯磷化合果糖6磷酸盐(f6p)实际应为标准物质中的α-gly,同理,可以得出,对比例2中的g1p、nuc均为错误的分析结果,实际分别应为标准化合物中的scyllo-ihp、β-gly。
综合来看,在样品单酯磷化合物错误解析的情况下,分析结果完全错误。如对比例2中31p-nmr谱图中单酯磷位移误判,导致一些峰找不到对应的物质,如谱图峰编号9对应的单酯磷化合物。再者,由于单酯磷化合物结构分析错误,单酯磷的含量也必然受到影响。如对比例2与实施例1获得的含量上差异很大,如由于把β-gly判断为nuc,其含量分析结果也由2.9mg/kg增加至9.1mg/kg,增加了近3倍。
上述例子仅作为说明的目的,本发明的范围并不受此限制。对本领域的技术人员来说进行修改是显而易见的,本发明仅受所附权利要求范围的限制。
- 还没有人留言评论。精彩留言会获得点赞!