评估高聚合物材料的能量损耗的方法与流程
本发明涉及一种精确的评估高聚合物材料的能量损耗的计算机化方法。
背景技术:
高聚合物材料例如塑料以及橡胶的能量损耗是非常重要的影响高聚合物材料产品的各种性能的物理量。例如,轮胎是一种典型的橡胶制品,并且其内使用的橡胶的能量损耗与该轮胎的抓地性能、燃料消耗性能等密切相关。因此精确地知道并控制能量损耗是很重要的。一般而言,例如,为了得到具有期望的物理量的橡胶组合物,一个研制周期——即设计橡胶组合物、试生产橡胶组合物、以及测定能量损耗将被重复进行直到能够获得期望的物理量。近年来,另一方面,例如,如下述专利文献1和2所公开的,已经提出根据高聚合物材料的计算机模拟的结果来评估高聚合物材料的能量损耗。[专利文献1]日本专利申请公开No.2007—107968[专利文献2]日本专利申请公开No.2009—271719专利文献1和2中公开的方法中,简略地说,以下述次序实施如下步骤(a)-(e):(a)定义高聚合物材料的分子结构模型,(b)在分子结构模型上定义位能,(c)使用高聚合物材料模型进行结构弛豫计算,其中分子结构模型被随机地设置在单元中,(d)结构弛豫计算之后,通过使高聚合物材料模型周期性变形来计算应力和应变,和(e)从计算结果得到滞后回线,并且通过滞后回线的面积计算能量损耗。然而,在这种方法中,存在如下问题:步骤(d)中,在计算期间,计算有时会不稳定并异常终止;以及在步骤(e)中,能量损耗的计算值有时与实际测量值相差很大。本发明人研究发现,当基于位能的特性频率确定的分子结构模型的振动周期变得与高聚合物材料模型的周期性变形的周期实质上相等时,计算变得不稳定并异常终止。
技术实现要素:
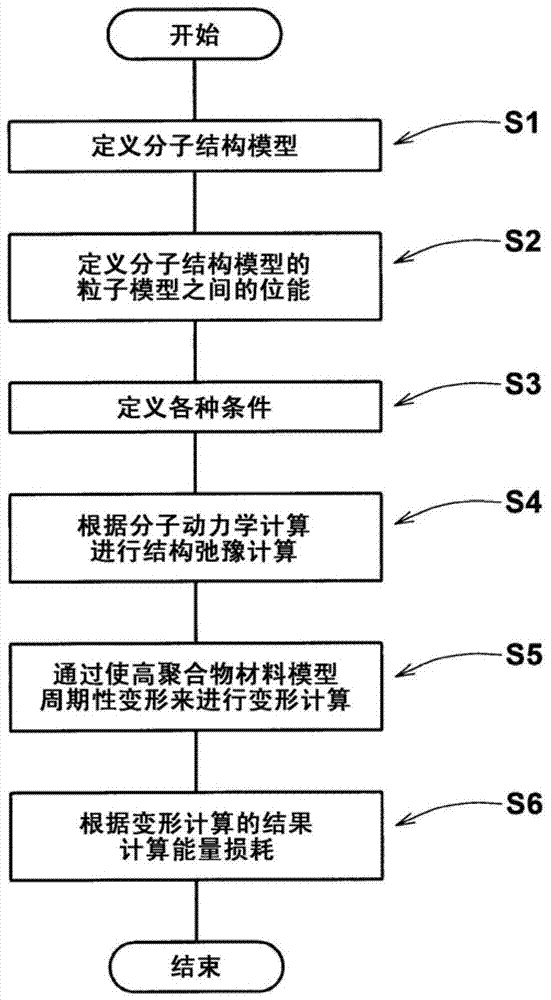
因此,本发明的目的在于提供一种用于评估高聚合物材料的能量损耗的计算机化方法,其中仅当分子结构模型的振动周期与高聚合物材料模型的周期性变形的周期不相等时才进行变形计算,由此可精确、稳定地评估高聚合物材料的能量损耗。根据本发明,用于评估高聚合物材料的能量损耗的计算机化方法包括:定义高聚合物材料的分子结构的分子结构模型的步骤,其中分子结构模型包括粒子模型和连接链,粒子模型代表原子或者原子集合体以及连接链用于确定粒子模型之间的相对位置,定义分子结构模型的粒子模型之间的位能的步骤,定义高聚合物材料模型以及对高聚合物材料模型进行结构弛豫计算的步骤,在高聚合物材料模型中,分子结构模型被设置于其单元中,以及结构弛豫计算基于位能和预定条件进行,进行用以获得应力和应变的变形计算的步骤,变形计算在进行结构弛豫计算之后,通过使高聚合物材料模型周期性变形来进行,基于应力和应变的变形计算的结果计算能量损耗的步骤,其中仅当周期性变形的周期Ts与基于位能的特有频率确定的振动周期Tp实质上不相等时,才进行变形计算。优选地,结构弛豫计算在这样的条件下进行:高聚合物材料模型的密度被定义为低于高聚合物材料的实际密度。优选地,结构弛豫计算在恒温、恒压下进行至少10皮秒。优选地,仅对其惯量半径少于0.5倍单元尺寸的分子结构模型进行变形计算。优选地,当周期Ts与振动周期Tp的比率Ts/Tp在0.8到1.2的范围之外时,进行变形计算。附图说明图1是聚丁二烯的结构式。图2是本发明实施方式的模拟方法的流程图。图3显示了实施方式中使用的分子结构模型。图4是聚合度为3的分子结构模型的透视图。图5是用于说明高聚合物材料模型的概略透视图。图6是显示分子结构模型的密度与结构弛豫计算的计算时间之间关系的曲线图。图7是实施方式中变形计算的流程图。图8(a)是用于说明变形计算的单元的示意图。图8(b)是用于说明应变变化的曲线图。图9是显示通过变形计算得到的应变和应力之间关系的曲线图。图10(a)和(b)是用于说明分子结构模型的惯量半径与单元尺寸之间关系的图。具体实施方式现在结合附图对本发明的实施方式进行详细说明。根据本发明,用于评估高聚合物材料的能量损耗的方法通过计算机完成。高聚合物材料至少包含橡胶、树脂以及弹性体。如图1所示,本实施方式中使用的高聚合物材料是顺式-1,4聚丁二烯(以下简称为聚丁二烯)。聚丁二烯是由甲撑基(-CH2-)以及次甲基(-CH-)形成的单体{-[CH2-CH=CH-CH2]-}聚合而成的聚合物。图2显示了本发明实施方式的模拟方法的流程图。在此实施方式中,首先高聚合物材料的分子结构模型通过计算机产生并被存储。(步骤S1)图3和图4显示分子结构模型2,其代表聚丁二烯的大分子链单元。当然,分子结构模型实际上是能够通过计算机用分子动力学计算处理的数据。分子结构模型2通过连接若干个(n个)丁二烯单体的单体模型3形成。在如图4所示的例子中,聚合度是3。本例中单体模型3是所谓的“全原子模型”,其中所有原子(C和H)在计算机模拟中作为粒子模型4被处理。然而,单体模型3也可能是所谓的“单原子模型”,其中多个原子被结合成一个粒子,其用一个粒子模型表示。在基于分子动力学计算的计算机模拟中,每个结合于分子结构模型2中的粒子模型4在运动方程式中作为质点被处理。换句话说,在计算机中定义并储存各个粒子模型4的参数数据-质量、体积、直径、电荷和/或初始坐标。连接链5被定义为使粒子模型4之间互相连接。连接链5是作为弹簧来处理的,并且为此,每个连接链5的平衡长度和弹簧常数被定义并存储于计算机中。每一单体模型3都是三维的。对于每一个单体模型3,粒子模型4之间的键长,各自由三个通过连接链5连接的粒子模型4形成的键角,以及各自通过由连接链5连接的四个粒子模型4中的三个相邻粒子模型4形成的二面角均被定义并存储在计算机中。当单体模型3受到外力或者内力时,其键长、键角以及二面角可能发生变化,于是,分子结构模型2的三维结构可能发生变化。这样的模型可以通过利用例如JSOL股份有限公司研制的“J-OCTA”软件处理。然后,粒子模型4之间的位能被定义并存储在计算机中。(步骤S2)位能是粒子之间的角度或距离的函数,并且用于计算作用于两个粒子模型4之间的力。主要的位能如下:通过连接链5连接的粒子模型4之间的键伸缩位能;三个连续的粒子模型4形成的弯曲位能;三个或四个连续的粒子模型4形成的扭转位能;以及不相互连接的粒子模型4之间的范德华力位能。然后,如图5所示,上述初始化的分子结构模型2被设置于预定单元(空间)S中,由此定义了高聚合物材料模型P。在此,分子结构模型2被随机地设置以形成稳定状态或亚稳定状态。然后,根据位能、预定的条件以及分子动力学计算进行结构弛豫计算。(步骤S3和S4)单元S相当于高聚合物材料的显微结构部分。在本例中,单元S是正六面体。在单元S中,聚合度为3的多个分子结构模型2被随机设置。结构弛豫计算的条件可以被设定为多种。在本实施方式中,高聚合物材料模型P的密度被设定成小于聚丁二烯的实际密度的值。此处,模型的密度是设置于单元S中的分子结构模型2的总质量除以单元S的体积的商。在本实施方式中,分子结构模型的密度默认被设定为0.01g/cm3。如果大量的分子结构模型2从开始就被设置于单元S中,那么当分子结构模型2相互重叠时,分子动力学计算就可能出现异常力,并且计算机的计算就变得不稳定。然而,在本实施方式中,由于结构弛豫计算在低密度条件下开始,分子结构模型2互相重叠的概率减少并且可以稳定计算。优选在恒压、恒温并在周期性边界条件下,结构弛豫计算进行至少10皮秒。作为结构弛豫计算结果的例子,图6显示了分子结构模型2的密度与计算时间之间的关系。如图所示,通过使结构弛豫计算进行至少10皮秒,高聚合物材料模型的密度恢复到其实际值。结果是,对高聚合物材料模型P进行的下一步变形计算变稳定。接着,通过将周期性变形施加到通过结构弛豫计算稳定的高聚合物材料模型P上来进行变形计算,以计算应力和应变。(步骤S5)图7显示了变形计算步骤S5的一个例子的流程图。在此例中,首先,基于对分子结构模型2定义的各个位能的特性频率,通过计算机计算振动周期Tp。(步骤S51)例如,就键伸缩位能来说,如果C-C键伸缩位能U通过下述表达式(1)定义:U=0.5×k×(x-x0)2---(1)其中,“k”是弹簧常数,“x”是C-C键长,“x0”是连接链的平衡长度,那么键伸缩的特性频率可以通过以下表达式(2)得到:f=1/(2pi)×(k/M)0.5---(2)其中,“pi”是圆周率,“M”是碳原子质量。振动周期Tp是特性频率的倒数。因此,键伸缩位能的特性频率f的振动周期Tp通过下述表达式(3)得到:Tp=1/f---(3)接着,设定高聚合物材料模型P的变形计算的周期Ts为某一个值。(步骤S52)如图8(a)所示,在本实施方式的变形计算中,模拟拉伸以及压缩变形,其中高聚合物材料模型P受到绝对值相同的单轴拉伸应变以及压缩应变,并且其从初始状态到初始状态(零应变到零应变)的过程被认为是一个周期。图8(b)显示了高聚合物材料模型P的周期性变形的例子。在图中,应变作为时间的函数被显示。在本例子中,应变像正弦波一样改变。它也可以像三角波一样改变。此外,除了周期Ts之外,还设定了应变的量级、泊松比等。接着,计算机断定周期Ts是否实质上等于振动周期Tp。(步骤S53)在多个不同的振动周期Tp存在的情况下,针对每一个振动周期Tp进行上述判断。在本实施方式中,如果比率Ts/Tp在0.8-1.2的范围之内,周期Ts就被认为实质上等于振动周期Tp。当然可以设定不同的范围。如果相等(步骤S53中的是),那么变形计算的周期Ts被调整到与前值不同的值。(步骤S52)如果不相等(步骤S53中的否),那么以周期Ts对高聚合物材料模型P进行变形计算。(步骤S54)如上所述,如果振动周期Tp实质上等于周期Ts,那么计算变得不稳定并异常终止。进一步地,计算结果的精确度大大降低。其理由被假定为,在变形的重复期间,原子之间距离的较大变化导致异常大的力作用在原子上。这样的问题似乎可以通过使变形计算仅进行一个周期来解决(即,不重复变形计算)。但是,当周期性变形通过分子动力学进行时,不管周期Tp是否等于特性频率Ts,在变形开始后,变形对应力的影响不能被识别或很难在短时间内识别。因此,使变形计算仅进行一个周期不是现实的方法。因此,在本发明中,在对高聚合物材料模型P进行变形计算之前,将高聚合物材料模型P的变形周期Ts与基于分子结构模型2的位能的特性频率确定的振动周期Tp相比较,并且如果周期Ts实质上不等于振动周期Tp,那么对高聚合物材料模型P进行变形计算。因此,在变形计算中,周期Ts永远不等于振动周期Tp。相应地,可以防止计算的异常终止以及计算精度的劣化。图9显示了通过变形计算得到的高聚合物材料模型P的应力-应变曲线(滞后回线)。高聚合物材料模型P的能量损耗可以通过计算滞后回线环绕的面积得到。优选变形计算仅针对其惯量半径少于0.5倍单元S的尺寸的分子结构模型进行。图10(a)显示惯量半径Rg大于0.5倍的单元S的尺寸L(这个例子中,单元S的边长)的例子。在使用分子结构模型的变形计算中,周期性边界条件被定义,其中单元S在前后方向、左右方向以及上下方向重复出现。在周期性边界条件中,一个单元S的边界紧靠相邻另一单元S的边界。如果第一个单元S中的分子结构模型2从第一个单元S的边界Sa中部分突出,那么与第一个单元的突出部分对应的在相邻的第二个单元S中的分子结构模型2的一部分从相对边界Sb突出到第一个单元S内。因此,如果惯量半径Rg大于单元S的尺寸L的0.5倍,那么分子结构模型的粒子模型可能互相重叠,于是,由于它们的位能而导致的异常力被计算。另一方面,如图10(b)所示,如果惯量半径Rg少于单元S的尺寸L的0.5倍,那么上述干扰或重叠可以被避免,并且可以进行精确计算。比较试验为了证实本发明的效果,依照上述实施方式进行能量损耗评估。条件如下:对应于键长的键伸缩位能的弹簧常数k=918[ε/σ2],粒子模型的质量M=1.0[m],特性频率f=4.82[1/τ],以及振动周期=0.207[τ],其中,ε:能量单位σ:长度单位,m:质量单位,以及τ:时间单位,在本实施方式中,变形的周期Ts被设定为2.07[τ],即是基于位能的特性频率确定的振动周期Tp的10倍。因此,可以得到如图9所示的如滞后回线的应力-应变曲线。作为对比,周期Ts被设定为0.207[τ],即等于振动周期的值,并进行计算。在这种情况下,计算异常结束。进一步地,可以确定,当比率Ts/Tp在0.8-1.2的范围内时,计算异常结束的概率较高。如上所述,在根据本发明的方法中,计算可以稳定地进行而不会异常结束,并且精确精算是可能的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1