循环肿瘤DNA重复序列的处理方法及装置与流程
本发明涉及遗传工程领域,具体而言,涉及一种循环肿瘤DNA重复序列的处理方法及装置。
背景技术:
:肿瘤细胞在进行分裂增殖过程当中,会凋亡、死亡、坏死,也会主动向体液中释放携带有肿瘤突变的DNA碎片,也即循环肿瘤DNA(circulatingtumorDNA,简称为ctDNA),多存在于血液、滑膜液和脑脊液等体液中,尤其是血浆游离DNA(cell-freeDNA,cfDNA)中。通过对ctDNA的测序,检测肿瘤细胞DNA分子上发生碱基序列改变(突变)的基因组区域,能够有效反应病人对治疗的响应;在检测到药物响应之后,肿瘤有可能对药物治疗产生耐药,ctDNA检测也可以追踪耐药突变的产生,定性定量;检测手术后是否存在残余组织,判断预后效果以及早期肿瘤的筛查。不同于常规的基因组DNA,ctDNA片段较短,通常只有100~400bp,而且在血液中含量较少,所以实际中能提取到的ctDNA量含量很低。由于ctDNA片段较短且含量较低的特点,因此在提取量较少时,需要在建库阶段进行多轮聚合酶链式反应(PolymeraseChainReaction,简称为PCR),扩大原始提取DNA的含量,以产生足够的DNA分子数目做高通量测序(High-throughputsequencing,简称为NGS测序)和后续生物信息学分析。由于PCR扩增导致对一个分子进行多次镜像复制,产生重复序列(Duplicatedreads),这些无效的重复数据对于检测变异极容易引入人工误差。对于最理想的NGS数据分析流程中,都需要尽可能把所有通过PCR获得的测序数据全部去除,还原到没有PCR的状态。现有技术中,提供了两种重复序列的去重方法,samtoolsrmdup和Picard’sMarkDuplicates。其中,samtoolsrmdup的工作原理为:NGS测序得到的序列(read)通过与人类参考基因组比对(mapping),得到这条read的比对位置,如果不同的reads比对到相同的基因组位置,则认为这部分的reads是通过PCR产生的多个重复序列,只保留mapping质量最高的read,删除其余的重复序列。对于PEreads,如果两端的read比多到基因组的不同染色体上或者两者之前的距离过长(即不是ProperPaired),则不作去重考虑。Picard’sMarkDuplicates的基本思路与samtoolsrmdup相同,通过比较reads中5'端的mapping位置,对于具有相同5'位置的序列,选取测序质量最高的reads作为去重后保留的唯一reads,且对于PEreads不是ProperPaired的情况也会做去重处理。但在基因组相同位置上,往往有可能会存在多个原始分子,这些原始分子并不是通常意义上的PCR重复,有可能存在有意义的突变(例如ctDNA中就是肿瘤相关变异),但在上述的去重方法中,对于这种情况的判断,samtoolsrmdup和Picard’sMarkDuplicates会错误的认为是同一个原始分子,仅保留1对reads,导致过度去重,浪费了部分有意义的数据量。无论是Samtoolsrmdup还是Picard’sMarkDuplicates都只考虑了read的某种质量值,而并没有考虑read上的具体序列上的差异,导致过去重或错误选取保留的uniquereads的发生。针对现有技术中测序数据的处理方法对样本测序进行重复序列删除或标记,准确度低的问题,目前尚未提出有效的解决方案。技术实现要素:本发明实施例提供了一种循环肿瘤DNA重复序列的处理方法及装置,以至少解决现有技术中测序数据的处理方法对样本测序进行重复序列删除或标记,准确度低的技术问题。根据本发明实施例的一个方面,提供了一种循环肿瘤DNA重复序列的处理方法,包括:获取待检测循环肿瘤DNA的测序数据和参考基因组序列,其中,测序数据为对待检测循环肿瘤DNA进行高通量测序得到的数据,测序数据包括:多对双端序列;将测序数据和参考基因组序列进行比对,得到第一比对结果,其中,第一比对结果至少包括:多对双端序列的基因组位置、碱基序列和对应的碱基质量值序列;基于第一比对结果,确定每对双端序列的类型,其中,类型包括:独立序列和重复序列。进一步地,基于第一比对结果,确定每对双端序列的类型包括:将多对双端序列划分为至少一个序列集合,其中,每个序列集合包括:至少一对双端序列,同一个序列集合中的双端序列的基因组位置相同且碱基序列相同;计算每个序列集合中每对双端序列包含的所有碱基的碱基质量值之和,得到每个序列集合中每对双端序列的碱基质量和;获取每个序列集合中最大碱基质量和对应的第一双端序列;将每个序列集合中第一双端序列作为独立序列,并将每个序列集合中除第一双端序列之外的其他第二双端序列作为重复序列。进一步地,将多对双端序列划分为至少一个序列集合包括:将每对双端序列的与多对双端序列中除每对双端序列之外的任意一对双端序列进行比较;如果每对双端序列的基因组位置和任意一对双端序列的基因组位置相同,并且每对双端序列中每个碱基位置上的碱基类型与任意一对双端序列中每个碱基位置上的碱基类型相同,则将每对双端序列和任意一对双端序列划分为同一个序列集合。进一步地,在基于第一比对结果,确定每对双端序列的类型之后,上述方法还包括:在第一比对结果中,对重复序列进行标记。进一步地,第一比对结果还包括:标记位,其中,在第一比对结果中,对重复序列进行标记包括:获取重复序列的标记位的当前值;计算当前值与预设值之和,得到和值;将当前值修改为和值。进一步地,在第一比对结果中,对重复序列进行标记之后,上述方法还包括:按照每对双端序列的基因组位置,显示第一比对结果。进一步地,在第一比对结果中,对重复序列进行标记之后,上述方法还包括:根据第一比对结果,显示每个基因组位置对应的双端序列的比对信息和碱基质量值;对比对质量满足预设条件的双端序列进行过滤。进一步地,在基于第一比对结果,确定每对双端序列的类型之前,上述方法还包括:按照每对双端序列的基因组位置,对第一比对结果进行排序,得到第二比对结果,并为第二比对结果建立索引;对第二比对结果进行过滤,得到第三比对结果;基于第三比对结果,确定每对双端序列的类型。进一步地,将测序数据和参考基因组序列进行比对,得到第一比对结果包括:获取多对双端序列中每条序列和参考基因组序列中的每段序列的匹配度;获取最高匹配度对应的至少一段序列,得到每条序列的匹配序列;根据每条序列的匹配序列,确定每条序列的基因组位置。根据本发明实施例的另一方面,还提供了一种循环肿瘤DNA重复序列的处理装置,包括:获取模块,用于获取待检测循环肿瘤DNA的测序数据和参考基因组序列,其中,测序数据为对待检测循环肿瘤DNA进行高通量测序得到的数据,测序数据包括:多对双端序列;比对模块,用于将测序数据和参考基因组序列进行比对,得到第一比对结果,其中,第一比对结果至少包括:多对双端序列的基因组位置、碱基序列和对应的碱基质量值序列;确定模块,用于基于第一比对结果,确定每对双端序列的类型,其中,类型包括:独立序列和重复序列。根据本发明实施例的另一方面,还提供了一种存储介质,存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行上述的循环肿瘤DNA重复序列的处理方法。根据本发明实施例的另一方面,还提供了一种处理器,处理器用于运行程序,其中,程序运行时执行上述的循环肿瘤DNA重复序列的处理方法。在本发明实施例中,获取待检测循环肿瘤DNA的测序数据和参考基因组序列,将测序数据和参考基因组序列进行比对,得到第一比对结果,进一步基于第一比对结果,确定每对双端序列的类型,从而实现重复序列的去重处理。容易注意到的是,由于在将测序数据和参考基因组序列进行比对之后,需要结合多对双端序列的具体碱基序列确定每对双端序列的类型,从而实现在考虑到序列质量值的同时,考虑具体碱基序列上的差异,达到保留更多的原始分子,提高处理准确度的技术效果,进而解决了现有技术中测序数据的处理方法对样本测序进行重复序列删除或标记,准确度低的技术问题。附图说明此处所说明的附图用来提供对本发明的进一步理解,构成本申请的一部分,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:图1是根据本发明实施例的一种循环肿瘤DNA重复序列的处理方法的流程图;图2是根据本发明实施例的一种可选的循环肿瘤DNA重复序列的处理方法的示意图;图3是根据本发明实施例的一种可选的循环肿瘤DNA重复序列的处理方法的流程图;以及图4是根据本发明实施例的一种循环肿瘤DNA重复序列的处理装置的示意图。具体实施方式为了使本
技术领域:
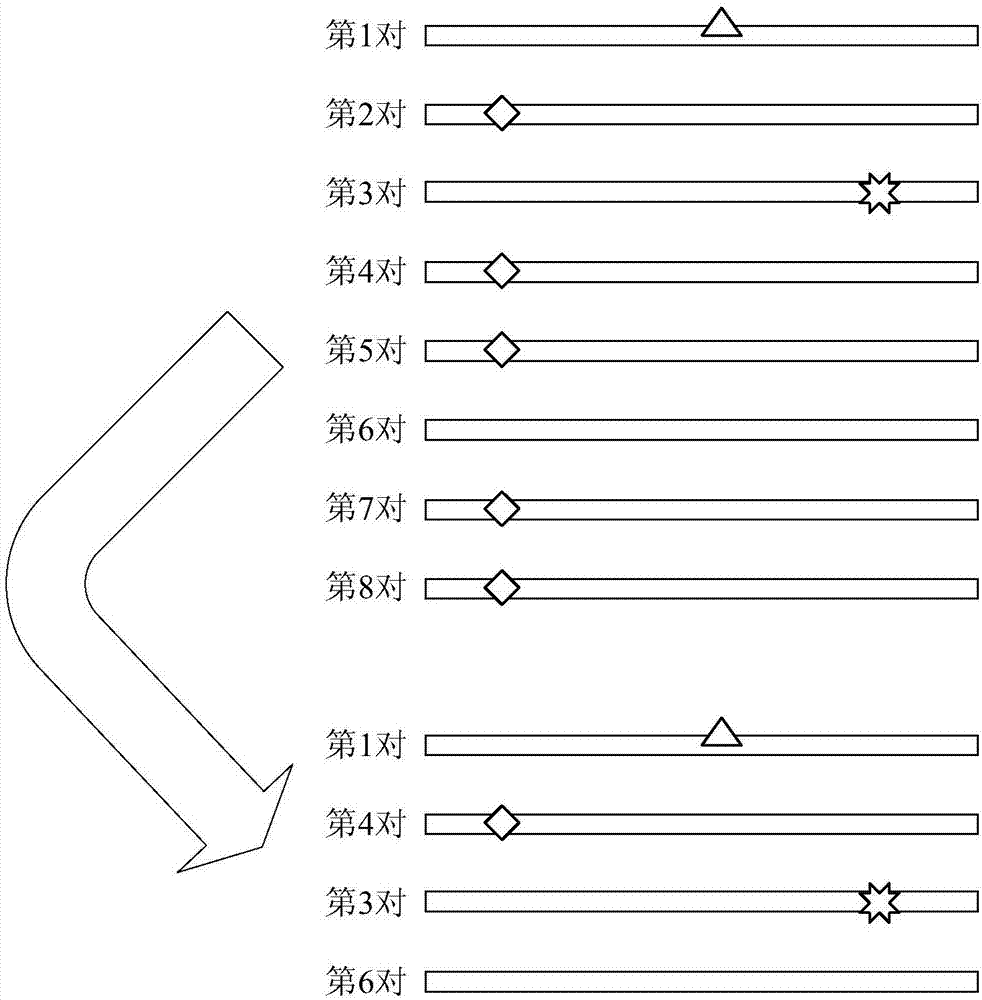
的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本发明的实施例能够以除了在这里图示或描述的那些以外的顺序实施。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。下面,首先对本发明实施例中出现的技术名词进行解释如下:NGS测序:高通量测序(High-throughputsequencing),又称“下一代”测序("Next-generation"sequencing,简称为NGS),是相对于传统的桑格测序(SangerSequencing)而言的,以能一次并行对几十万到几百万条DNA分子进行序列测定和一般读长较短等特点为标志。NGS技术目前已经在诊断遗传病、测量基因表达水平、构建进化树、区分形态学上近似物种、对非模式生物进行重头测序头(denovosequencing),构建其参考基因组序列等多个领域得到广泛应用。NGS测序有单端(Single-end,简称为SE)和双端(Paird-end,PE)测序之分,目前大部分使用的都是PE测序技术,PE即指同一个DNA分子的两头,测序时先测一端,然后反过来再测另一端。测的这两条序列就叫做PairedendsReads(PEReads),很多时候,它又被叫做"MatePaires"。捕获测序:将感兴趣的基因组区域定制成特异性探针与基因组DNA在序列捕获芯片(或溶液)进行杂交,将目标基因组区域的DNA片段进行富集后再利用NGS测序技术进行测序的研究策略。由于其捕获区域短,大大降低测序成本,被各领域广泛应用,肿瘤检测领域也多使用捕获测序的策略。ctDNA:循环肿瘤DNA,肿瘤细胞在进行分裂增值过程当中,主动向体液中分泌的已经经历过基因突变的DNA片段。PCR:聚合酶链式反应,一种用于放大扩增特定的DNA片段的技术。重复序列:由于PCR扩增导致对一个分子进行多次镜像复制的后果。reads:测序读长,测序仪测到的基因组或转录组序列片段。fasta:一种基于文本用于表示核苷酸序列或氨基酸序列的格式。fastq:一种常见的高通量测序文件类型,通常原始测序数据都是以该文件类型储存的。bwa:一种比对方法软件,用于查找测序序列在人类基因参考序列中的位置,可输出bam格式结果文件。sam:一种序列比对格式,用来存储测序序列回贴到参考基因组的结果bam:sam文件的二进制压缩格式,用来存储测序序列回贴到参考基因组的结果。samtools:一种处理bam/sam文件的工具。picard:一种处理高通量测序数据的工具,可用于处理sam/bam等比对结果文件。比对质量:用于量化比对到错误位置的可能性,值越高表示可能性越低。CIGAR:简要比对信息表达式,其以参考序列为基础,使用数据加字母表示比对结果。实施例1根据本发明实施例,提供了一种循环肿瘤DNA重复序列的处理方法的实施例,需要说明的是,在附图的流程图示出的步骤可以在诸如一组计算机可执行指令的计算机系统中执行,并且,虽然在流程图中示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。图1是根据本发明实施例的一种循环肿瘤DNA重复序列的处理方法的流程图,如图1所示,该方法包括如下步骤:步骤S102,获取待检测循环肿瘤DNA的测序数据和参考基因组序列,其中,测序数据为对待检测循环肿瘤DNA进行高通量测序得到的数据,测序数据包括:多对双端序列。具体地,上述的待检测循环肿瘤DNA可以从病人的血液、淋巴液、组织间隙液、脑髓液等体液中提取得到的基因序列,在本发明实施例中以血液中提取到的ctDNA为例进行说明;上述的测序数据可以是对待检测ctDNA进行NGS测序得到的ctDNA样本捕获测序fastq数据;上述的参考基因组序列可以是从公开数据库NCBI等网站下载的人类参考基因组fasta数据。步骤S104,将测序数据和参考基因组序列进行比对,得到第一比对结果,其中,第一比对结果至少包括:多对双端序列的基因组位置、碱基序列和对应的碱基质量值序列。具体地,上述的基因组位置可以是每对PEreads比对到参考基因组序列中的位置,不同的PEreads可以比对到相同的位置;上述的碱基质量值可以是通过NGS测序得到的测序质量,用于衡量每个碱基位置上碱基类型测量的准确度,碱基质量值越大,说明碱基类型测量的准确度越高;上述的碱基序列可以是每对双端序列中每个碱基位置上的碱基类型,DNA序列中包含四种类型的碱基,分别为G、C、T、A,在NGS测序过程中,可以确定每个碱基位置上的碱基类型,并得到该碱基类型的碱基质量值。在一种可选的方案中,可以获取人类参考基因组fasta数据和ctDNA样本捕获测序fastq数据,利用基因组比对工具bwamem进行序列比对,得到比对结果文件(.bam),也即,得到上述的第一比对结果,比对结果文件为bam格式,包含每对PEreads的名称、位置信息、SAM标记、比对质量信息、CIGAR字串、matepair信息、片段序列、测序质量等。需要说明的是,第一比对结果中的多对双端序列的碱基序列和碱基质量值是从NGS测序的测序数据中直接继承过来的数据,第一比对结果包含基因组比对位置及比对情况的信息的同时,还存储了多对双端序列的碱基序列和碱基质量值,方便后续的其他分析,不再使用fastq文件。步骤S106,基于第一比对结果,确定每对双端序列的类型,其中,类型包括:独立序列和重复序列。在一种可选的方案中,由于在没有PCR和测序错误时,同一个DNA分子片段(fragment)经过PCR产生多个完全一样的fragment,这组fragment均可以比对到参考基因组同一个位置,且序列相同。而来自不同fragment的DNA分子,虽然可能比对到基因组的相同位置,但由于其可能分别属于不同的DNA分子(例如,血液中提取到的DNA包含两种类型,一种是包含肿瘤信息的ctDNA分子;另一种是在血液中游离的自身DNA,多是从身体的细胞或者白血球破裂释放出来的,一般认为是无害的,不用多久会被人体自身清理掉,两种DNA携带的信息不同;又例如,不同的ctDNA分子),所以其序列可能并不相同。因此,本发明实施例提供了一种UniqS方法,基于上述基本事实,可以将比对到基因组相同位置且序列完全相同的PEreads定义为来自同一个原始fragment的多个重复,并选择其中碱基质量和最高的作为这组fragment的最终代表的独立序列(uniquereads),其余作为重复序列。需要说明的是,无论是通过本发明提供的去重方法进行去重之前,还是通过本发明提供的去重方法进行去重之后,所有的bam文件均包含下列信息:每对PEreads的名称、位置信息、SAM标记、比对质量信息、CIGAR字串、matepair信息、片段序列、测序质量等。根据本发明上述实施例,获取待检测循环肿瘤DNA的测序数据和参考基因组序列,将测序数据和参考基因组序列进行比对,得到第一比对结果,进一步基于第一比对结果,确定每对双端序列的类型,从而实现重复序列的去重处理。容易注意到的是,由于在将测序数据和参考基因组序列进行比对之后,需要结合多对双端序列的具体碱基序列确定每对双端序列的类型,从而实现在考虑到序列质量值的同时,考虑具体碱基序列上的差异,达到保留更多的原始分子,提高处理准确度的技术效果,进而解决了现有技术中测序数据的处理方法对样本测序进行重复序列删除或标记,准确度低的技术问题。可选地,在本发明上述实施例中,步骤S106,基于第一比对结果,确定每对双端序列的类型包括:步骤S1062,将多对双端序列划分为至少一个序列集合,其中,每个序列集合包括:至少一对双端序列,同一个序列集合中的双端序列的基因组位置相同且每个碱基位置上的碱基类型相同。具体地,上述的每个序列集合用于表征同一个fragment,每个序列集合中包含的PEreads为同一个fragment经过PCR产生的多个重复序列。步骤S1064,计算每个序列集合中每对双端序列包含的所有碱基的碱基质量值之和,得到每个序列集合中每对双端序列的碱基质量和。步骤S1066,获取每个序列集合中最大碱基质量和对应的第一双端序列。步骤S1068,将每个序列集合中第一双端序列作为独立序列,并将每个序列集合中除第一双端序列之外的其他第二双端序列作为重复序列。在一种可选的方案中,可以将比对到基因组相同位置且序列完全相同的PEreads定义为来自同一个原始fragment的多个重复,选择其中碱基质量和最高的作为这组fragment的最终代表的uniquereads,其余作为其副本标记为重复。例如,如图2所示,对于包含如2所示的8对PEreads的测序数据,可以将第1对PEreads划分为第一个序列集合,将第2对、第4对、第5对、第7对和第8对PEreads划分为第二个序列集合,将第3对PEreads划分为第三个序列集合,将第6对PEreads划分为第四个序列集合,从而得到四个序列集合,也即,测序数据中的8对PEreads来自四个fragment的多个重复,进一步地,对于第一个序列集合、第三个序列集合和第四个序列集合,由于序列集合中仅包含一对PEreads,因此,可以将序列集合中包含的PEreads作为uniquereads;对于第二个序列集合,由于序列集合中包含5对PEreads,则可以获取碱基质量和最高的PEreads,例如,第4对PEreads作为uniquereads,并将第2对、第5对、第7对和第8对PEreads作为重复序列进行标记。可选地,在本发明上述实施例中,步骤S1062,将多对双端序列划分为至少一个序列集合包括:步骤S10622,将每对双端序列的与多对双端序列中除每对双端序列之外的任意一对双端序列进行比较。步骤S10624,如果每对双端序列的基因组位置和任意一对双端序列的基因组位置相同,并且每对双端序列中每个碱基位置上的碱基类型与任意一对双端序列中每个碱基位置上的碱基类型相同,则将每对双端序列和任意一对双端序列划分为同一个序列集合。在一种可选的方案中,为了判断两对PEreads是否来自同一个fragment,则可以首先判断两对PEreads的基因组位置是否相同,如果不同,则可以确定两对PEreads来自不同的fragment,如果相同,则进一步判断两对PEreads每个碱基位置上的碱基类型是否相同,如果不同,则可以确定两对PEreads来自不同的fragment;如果相同,则可以确定两对PEreads序列完全相同,也即两对PEreads来自同一个fragment,因此,可以将两对PEreads划分在同一个序列集合中。可选地,在本发明上述实施例中,在步骤S106,基于第一比对结果,确定每对双端序列的类型之后,该方法还包括:步骤S108,在第一比对结果中,对重复序列进行标记。在一种可选的方案中,本发明提供的UniqS方法不删除原始数据,仅在比对结果文件(.bam)中对重复序列进行标记,得到标记后的比对结果文件(.bam)。可选地,在本发明上述实施例中,第一比对结果还包括:标记位,其中,步骤S108,在第一比对结果中,对重复序列进行标记包括:步骤S1082,获取重复序列的标记位的当前值。具体地,上述的标志位可以是比对结果文件(.bam)中的SAM标记,SAM标记的值可以是多种比对信息对应值之和,上述的当前值可以是SAM标记的原有数值,不同的PEreads的SAM标记的原有数值不同。步骤S1084,计算当前值与预设值之和,得到和值。具体地,上述的预设值可以是1024。步骤S1086,将当前值修改为和值。在一种可选的方案中,本发明提供的UniqS方法不删除原始数据,仅在相应的SAM标记的原有数值上增加1024,表示该PEreads为重复序列,从而得到标记后的比对结果文件(.bam)。可选地,在本发明上述实施例中,在步骤S108,在第一比对结果中,对重复序列进行标记之后,该方法还包括:步骤S110,根据第一比对结果,显示每个基因组位置对应的双端序列的比对信息和碱基质量值。具体地,上述的比对信息可以是是否与参考基因组的每个位置上的碱基相同,是否发生插入缺失,比对到参考基因组的正链还是负链等,本发明对此不作具体限定。步骤S112,对比对质量满足预设条件的双端序列进行过滤。具体地,上述的预设条件可以是比对质量为0。在一种可选的方案中,可以调用samtools-1.3mpileup根据标记后的bam文件,按基因组位置展示比对到该位置的所有reads的比对信息和质量值,每一行记录一个基因组位置的比对情况,也即,每一行记录比对到每个基因组位置的所有reads的比对信息和质量值。可以认为是一种比对结果纵向的堆叠式的统计和展示。可选地,在本发明上述实施例中,在步骤S108,在第一比对结果中,对重复序列进行标记之后,该方法还包括:步骤S114,获取捕获测序区间。步骤S116,根据捕获测序区间,对独立序列进行单核苷酸变异检测和插入缺失检测,得到检测结果。在一种可选的方案中,可以获取捕获测序区间Bed文件,并调用varscan2mpileup2snp模块检测单核苷酸变异(SNV),mpileup2indel模块检测插入缺失(INDEL),其中,单核苷酸变异是指参考基因组的某个位置上发生碱基类型的改变,插入缺失是指在参考基因组的某段序列上插入了一小段新的序列或缺失了某段序列。可选地,在本发明上述实施例中,在步骤S106,基于第一比对结果,确定每对双端序列的类型之前,该方法还包括:步骤S118,按照每对双端序列的基因组位置,对第一比对结果进行排序,得到第二比对结果,并为第二比对结果建立索引。在一种可选的方案中,可以调用Picard’sSortSam模块将比对结果文件(.bam)(也即上述的第一比对结果)按比对位置排序,同时建立bam文件的索引文件(.bai)。通过比对结果文件按比对位置排序,从而使得相同位置的PEreads相邻,方便后续对PEreads进行去重处理。步骤S120,对第二比对结果进行过滤,得到第三比对结果。在一种可选的方案中,由于同一个PEreads可能会对比到多个基因组位置,在进行去重处理之前,首先需要对比对结果文件(.bam)进行过滤,具体可以调用samtoolsview模块对排序后的bam文件进行筛选,得到第三比对结果。步骤S122,基于第三比对结果,确定每对双端序列的类型。在一种可选的方案中,可以将第三比对结果中,比对到基因组相同位置且序列完全相同的PEreads定义为来自同一个原始fragment的多个重复,并选择其中碱基质量和最高的作为这组fragment的最终代表的独立序列(uniquereads),其余作为重复序列。可选地,在本发明上述实施例中,步骤S104,将测序数据和参考基因组序列进行比对,得到第一比对结果包括:步骤S1042,获取多对双端序列中每条序列和参考基因组序列中的每段序列的匹配度。步骤S1044,获取最高匹配度对应的至少一段序列,得到每条序列的匹配序列。步骤S1046,根据每条序列的匹配序列,确定每条序列的基因组位置。在一种可选的方案中,可以计算每一对PEreads中每条reads与人类参考基因组序列的匹配度,通过匹配度判断每一条reads是否来自人类参考基因组序列中某一段序列,匹配度越高,每一条reads来自人类参考基因组序列中该序列的可能性越大,可以将每条reads比对到最高匹配度的序列,从而根据该序列的位置,可以得到该条reads的基因组位置。需要说明的是,在本发明实施例中,可以采用现有技术中提供的比对算法进行比对,本发明对此不做具体限定。图3是根据本发明实施例的一种可选的循环肿瘤DNA重复序列的处理方法的流程图,下面结合图3对本发明一种优选的实施例进行详细说明。如图3所示,该方法可以包括如下步骤:输入cfDNA样本捕获测序fastq文件和人类参考基因组fasta文件,利用bwamem软件进行基因组比对;调用Picard软件进行reads排序;调用samtools软件进行reads过滤;利用本发明上述实施例提供的UniqS算法进行去重,得到cfDNA样本标记重复后的bam文件;输入捕获测序区间Bed文件,调用samtoolsmpileup对标记重复后的bam文件按位置展示所有reads的比对情况和质量值;调用varscan2mpileup2snp模块鉴定SNV,mpileup2indel模块鉴定INDEL。需要说明的是,上述的cfDNA样本也可以是其他含有ctDNA的体液样本。本发明输入文件包括:待测样本经过比对、排序、过滤等步骤后生成的测序数据文件(bam格式,包含每条测序片段的名称、SAM标记、位置信息、比对质量信息、CIGAR字串、matepair信息、片段序列、测序质量等)、人类参考基因组序列(fasta格式);本发明的输出文件包括:待测样本标记重复后的比对结果文件(bam格式)以及检测到的SNV和INDEL的vcf格式文件。通过上述方案,对于DNA分子碎片化严重、覆盖基因组范围小、经过多轮PCR的样本或测序方案,尤其是血浆ctDNA样本的捕获测序数据可以保留更多的原始分子,有效利用碱基序列,提高了原始数据的利用率,和最终变异检测的准确性。下面通过单碱基变异(SNV)梯度稀释细胞系测试实验验证对上述实施例进行验证。1、细胞系培养细胞系HCT116、KYSE450、NCI-H1573、NCI-H1975、NCI-H441、PC-9、SK-HEP-1、SW48、THP-1、BEAS-2B购买自南京科佰生物科技有限公司,按照提供的说明书进行细胞培养,即RPMI-1640培养基中加入10%胎牛血清,在37度条件下进行培养。2、细胞DNA提取收集细胞悬液后,常温300g离心5分钟后弃上清,用200uLPBS重悬细胞,然后用QIAampDNAMiniKit(货号为51304;Qiagen,Germany)进行基因组DNA提取。经过裂解后过柱纯化,最后用low-TE缓冲液洗脱DNA。3、用ddPCR的方法确定以上细胞系中突变位点的理论VAF用细胞提取的基因组DNA作为模板,进行ddPCR的实验,以上细胞系中突变位点的理论VAF如表1所示。ddPCR用伯乐的仪器、商品化探针和反应体系。反应体系组成为:10ulddPCRsupermixforprobes(nodUTP),1ul突变探针,1ul野生型探针,以及20ng待测DNA。配制好反应体系后,按照仪器使用方法进行乳糜生成,吸取乳糜至96孔PCR板,用PierceableFoilHeatSeal进行热封。PCR反应的条件为:酶激活95度,8min;94度30s解链,55度1min退火延伸,共39个循环;酶失活98度10min;4度保温。PCR扩增之后,伯乐的微滴读取仪读取每个反应孔中的带有荧光的微滴数目。每批次反应用超纯水作为阴性对照。每个待测DNA做三个复孔作为技术重复。表14、含有11个突变位点的样本制备按照下表2中的质量百分比混合上表中的10种细胞系,制备成1个样本,并计算预期的VAF值。表25、样本的ddPCR结果用ddPCR实验的方法检测样本中以上列表中各个位点的VAF值,如表3所示,每个反应体系中加入20ng样本DNA,每个样本做三个复孔作为技术重复。表3基因突变DDPCRVAFKRASG13D0.53PIK3CAH1047R1.06EGFRG719S0.88NRASQ61K1.80EGFRL858R1.26EGFRT790M1.52KRASG12V1.43EGFRE746_A750del4.76BRAFV600E0.92EGFRS768I2.42NRASG12D4.486、样本的文库构建、捕获和测序将混合的细胞系样本DNA首先用covaris超声打断成200bp左右的DNA片段,qubit荧光定量后,如表4所示,用不同的起始量DNA,不足50ul用无酶水补平,采用KAPAhyperpreparationkit(罗氏公司)进行文库构建,经过末端修复、3’端加polyA、连接测序接头、进行无偏向扩增,之后进行纯化获得文库。表4样本起始量DNA(ng)PCR循环数样本1206样本258样本358详述如下:1)末端平齐并在3’末端加A:反应体系如下表5所示:表5试剂体积Fragmented,double-strandedDNA50μLEndRepair&A-TailingBuffer7μLEndRepair&A-TailingEnzymeMix3μL总体积60μLBuffer和酶应预先在EP管中混匀,与DNA涡旋混匀后按以下反应进行。反应步骤如下表6所示:表6该步操作将PCR管盖温度设为85℃,而非105℃。若该操作结束后立即进行下步实验,应将终止温度设为20℃,而非4℃。2)连接接头:根据建库说明书的指导,20ngDNA应该采用7.5uM接头。按照下表7所示配制反应体系:表7试剂体积反应产物60μL接头体积5μL超纯水5μL连接Buffer30μLDNA连接酶10μL总体积110μLBuffer和酶应预先在EP管中混匀,涡旋震荡后离心,20℃孵育15分钟。3)连接后纯化:在上一步反应体系(110ul)中加入AgencourtAMPureXP纯化磁珠88ul。充分涡旋振荡,轻微离心。室温吸附5-15分钟,使DNA与磁珠充分结合EP管放至磁力架吸附至液体澄清缓慢吸取EP管中上清并丢弃。EP管中加入200μL80%乙醇孵育30秒缓慢吸取EP管中乙醇并丢弃。重复一次乙醇洗磁珠。EP管室温干燥3-5分钟至乙醇完全挥发。从磁力架取下EP管,加入22μL超纯水,涡旋振荡,轻微离心室温孵育2分钟洗脱DNA,EP管放至磁力架吸附至液体澄清,上清转移至新的EP管,取1μL上清测DNA浓度,剩余的进行扩增。4)PCR扩增:按照下表8所示配制PCR体系。表8试剂体积KAPAHiFiHotStartReadyMix(2X)25μLKAPALibraryAmplificationPrimerMix(10X)*5μL接头连接文库20μL总体积50μL充分震荡后快速离心,按照下表9所示条件进行PCR反应。表95)扩增后纯化:加入与PCR反应体系同等体积的AgencourtAMPureXP纯化磁珠(50μl)。充分涡旋振荡,轻微离心,室温吸附5-15分钟,使DNA与磁珠充分结合。EP管放至磁力架吸附至液体澄清,缓慢吸取EP管中上清并丢弃。EP管中加入200μL80%乙醇孵育30秒,缓慢吸取EP管中乙醇并丢弃。重复一次乙醇洗磁珠。EP管室温干燥3-5分钟至乙醇完全挥发。从磁力架取下EP管,加入52μL超纯水,涡旋振荡,轻微离心。室温孵育2分钟洗脱DNA,EP管放至磁力架吸附至液体澄清,上清转移至新的EP管,取1μL上清测DNA浓度。6)在测序前采用探针捕获的方法,用RocheNimbleGen探针将包含11个突变位点的目的区域进行富集和进一步扩增,获得目的区域的文库。经过q-PCR定量后进行上机测序。7、处理下机fastq数据为各软件可使用的输入文件。数据下机后,首先将下机数据从fastq文件处理成bam文件,具体使用的软件和步骤如下:7.1比对调用bwa-0.7.12mem将每一对fastq文件都作为PEreads比对到hg19人类参考基因组序列,除-M参数与指定ReadsGroup的ID外,不使用其余参数选项,生成初始bam文件。7.2排序调用picard-2.1.0的SortSam模块,对初始bam文件按照染色体位置进行排序,参数设置为“SORT_ORDER=coordinate”。7.3筛选调用samtools-1.3view对排序后的bam文件进行筛选,采用“-F0x900”作为参数。7.4建立索引调用samtools-1.3的index模块对最终生成的bam文件建立索引,生成与过滤后的bam文件配对的bai文件。8、标记重复8.1使用Picard’sMarkDuplicates模块标记重复,后续的变异检测时,会自动过滤这部分重复序列,再进行分析。8.2根据本发明上述实施例提供的方法(UniqS)对过滤后的bam文件去除重复序列,生成标记重复的bam文件。8.3统计比对情况:调用samtools-1.3的flagstat模块对最终生成的bam文件进行统计,生成标记重复后的bam文件的比对情况文件,包括总reads的数量、重复reads的数量、比对到参考基因组上的reads数量、成对的reads数据数量、read1的数量、read2的数量、完美匹配到参考序列的reads数量(properlypaired)、一对reads都比对到了参考序列上的数量、一对reads中只有一条与参考序列相匹配的数量、一对reads比对到不同染色体的数量、一对reads比对到不同染色体的且比对质量值大于5的数量等。8.4结果比较:本发明上述实施例提供的算法与Picard方法的数据量统计结果如下表10所示,从下表10中可以看出,本发明提供的算法比Picard方法保留的数据量更多,提高了数据的有效利用率。表10样本PicardUniqS样本12487274758481983样本21368762655455207样本314290322526310439、变异检测9.1堆叠调用samtools-1.3mpileup对标记重复后的bam文件按位置展示所有reads的比对情况和质量值,参数设置为“q=1”,mpileup的结果文件(mpileup文件)包含染色体、基因组位置、参考基因组碱基类型、该位点测序深度、全部覆盖该位点reads的比对情况和质量值。由于ddPCR验证阳性位点有限,仅对下列区间做mpileup处理,使用参数“-lpositive.bed”,positive.bed文件如表11所示。表11染色体起始位置结束位置基因chr1115256527115256530NRASchr1115258745115258748NRASchr3178952083178952086PIK3CAchr122539827925398282KRASchr122539828225398285KRASchr7140453134140453137BRAFchr75524170655241709EGFRchr75524241455242513EGFRchr75524900355249006EGFRchr75524906955249072EGFRchr75525951355259516EGFR9.2统计positive.bed区间的平均测序深度使用简单的脚本或bash命令根据mpileup文件统计不同去除重复序列方法在positive.bed区间的测序深度的平均值,结果见表12。表12样本PicardUniqS样本11625.3704524.840样本2533.4963855.390样本3627.3803601.530本发明提供的方法比Picard的方法在positive.bed区间平均深度相比Picard去重更高。9.3变异检测调用varscan2mpileup2snp模块检测单核苷酸变异(SNV),mpileup2indel模块检测插入缺失标记(INDEL),参数设置“--min-coverage100--min-reads22--min-var-freq0.001--p-value0.05--min-avg-qual20”。对上述3个样本的ddPCR验证为阳性的位点用不同去重方法之后统计的变异结果如下表13至15所示(表格中数值为突变频率),其中,表13示出样本1的变异结果,表14示出样本2的变异结果,表15示出样本3的变异结果。表13基因AachangePicardUniqSNRASp.Q61K01.05PIK3CAp.H1047R0.961.15BRAFp.V600E0.830.73NRASp.G12D3.874.8EGFRp.G719S0.880.74EGFRp.L858R1.641.93EGFRp.S768I2.152.33KRASp.G13D0.60.5EGFRp.745_750del3.052.69KRASp.G12V1.021.15EGFRp.T790M1.391.04表14基因AachangePicardUniqSNRASp.Q61K4.224.23PIK3CAp.H1047R01.77BRAFp.V600E00.92NRASp.G12D01.53EGFRp.G719S0.931.3EGFRp.L858R2.32.35EGFRp.S768I1.040.83KRASp.G13D1.070.86EGFRp.745_750del2.922.03KRASp.G12V1.341.83EGFRp.T790M0.960.87表15基因AachangePicardUniqSNRASp.Q61K00.99PIK3CAp.H1047R00.5BRAFp.V600E0.990.94NRASp.G12D5.455.83EGFRp.G719S01.32EGFRp.L858R0.760.94EGFRp.S768I1.661.7KRASp.G13D00.3EGFRp.745_750del2.562.35KRASp.G12V1.542.04EGFRp.T790M00.88Picard在多处阳性位点检测的突变频率为0(频率>0为阳性,频率=0为阴性),而UniqS方法在全部11个位点都检测为阳性。综上可以看出使用本发明相比Picard去重可以检测更多的阳性位点。实施例2根据本发明实施例,提供了一种循环肿瘤DNA重复序列的处理装置的实施例。图4是根据本发明实施例的一种循环肿瘤DNA重复序列的处理装置的示意图,如图4所示,该装置包括:获取模块42,用于获取待检测循环肿瘤DNA的测序数据和参考基因组序列,其中,测序数据为对待检测循环肿瘤DNA进行高通量测序得到的数据,测序数据包括:多对双端序列。具体地,上述的待检测循环肿瘤DNA可以从病人的血液、淋巴液、组织间隙液、脑髓液等体液中提取得到的基因序列,在本发明实施例中以血液中提取到的ctDNA为例进行说明;上述的测序数据可以是对待检测ctDNA进行NGS测序得到的ctDNA样本捕获测序fastq数据;上述的参考基因组序列可以是从公开数据库NCBI等网站下载的人类参考基因组fasta数据。比对模块44,用于将测序数据和参考基因组序列进行比对,得到第一比对结果,其中,第一比对结果至少包括:多对双端序列的基因组位置、碱基序列和对应的碱基质量值序列。具体地,上述的基因组位置可以是每对PEreads比对到参考基因组序列中的位置,不同的PEreads可以比对到相同的位置;上述的碱基质量值可以是通过NGS测序得到的测序质量,用于衡量每个碱基位置上碱基类型测量的准确度,碱基质量值越大,说明碱基类型测量的准确度越高;上述的碱基序列可以是每对双端序列中每个碱基位置上的碱基类型,DNA序列中包含四种类型的碱基,分别为G、C、T、A,在NGS测序过程中,可以确定每个碱基位置上的碱基类型,并得到该碱基类型的碱基质量值。在一种可选的方案中,可以获取人类参考基因组fasta数据和ctDNA样本捕获测序fastq数据,利用基因组比对工具bwamem进行序列比对,得到比对结果文件(.bam),也即,得到上述的第一比对结果,比对结果文件为bam格式,包含每对PEreads的名称、位置信息、SAM标记、比对质量信息、CIGAR字串、matepair信息、片段序列、测序质量等)。需要说明的是,第一比对结果中的多对双端序列的碱基序列和碱基质量值是从NGS测序的测序数据中直接集成过来的数据,第一比对结果包含基因组比对位置及比对情况的信息的同时,还存储了多对双端序列的碱基序列和碱基质量值,方便后续的其他分析,不再使用fastq文件。确定模块46,用于基于第一比对结果,确定每对双端序列的类型,其中,类型包括:独立序列和重复序列。在一种可选的方案中,由于在没有PCR和测序错误时,同一个DNA分子片段(fragment)经过PCR产生多个完全一样的fragment,这组fragment均可以比对到参考基因组同一个位置,且碱基序列相同。而来自不同fragment的DNA分子,虽然可能比对到基因组的相同位置,但由于其可能分别属于不同的DNA分子(例如,血液中提取到的DNA包含两种类型,一种是包含肿瘤信息的ctDNA分子;另一种是在血液中游离的自身DNA,多是从身体的细胞或者白血球破裂释放出来的,一般认为是无害的,不用多久会被人体自身清理掉,两种DNA携带的信息不同;又例如,不同的ctDNA分子),所以其序列可能并不相同。因此,本发明实施例提供了一种UniqS方法,基于上述基本事实,可以将比对到基因组相同位置且序列完全相同的PEreads定义为来自同一个原始fragment的多个重复,并选择其中碱基质量和最高的作为这组fragment的最终代表的独立序列(uniquereads),其余作为重复序列。需要说明的是,无论是通过本发明提供的去重方法进行去重之前,还是通过本发明提供的去重方法进行去重之后,所有的bam文件均包含下列信息:每对PEreads的名称、位置信息、SAM标记、比对质量信息、CIGAR字串、matepair信息、片段序列、测序质量等。根据本发明上述实施例,获取待检测循环肿瘤DNA的测序数据和参考基因组序列,将测序数据和参考基因组序列进行比对,得到第一比对结果,进一步基于第一比对结果,确定每对双端序列的类型,从而实现重复序列的去重处理。容易注意到的是,由于在将测序数据和参考基因组序列进行比对之后,需要结合多对双端序列的具体碱基序列确定每对双端序列的类型,从而实现在考虑到序列质量值的同时,考虑具体碱基序列上的差异,达到保留更多的原始分子,提高处理准确度的技术效果,进而解决了现有技术中测序数据的处理方法对样本测序进行重复序列删除或标记,准确度低的技术问题。实施例3根据本发明实施例,提供了一种存储介质的实施例,存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行上述的循环肿瘤DNA重复序列的处理方法。实施例4根据本发明实施例,提供了一种处理器的实施例,处理器用于运行程序,其中,程序运行时执行上述的循环肿瘤DNA重复序列的处理方法。上述本发明实施例序号仅仅为了描述,不代表实施例的优劣。在本发明的上述实施例中,对各个实施例的描述都各有侧重,某个实施例中没有详述的部分,可以参见其他实施例的相关描述。在本申请所提供的几个实施例中,应该理解到,所揭露的技术内容,可通过其它的方式实现。其中,以上所描述的装置实施例仅仅是示意性的,例如所述单元的划分,可以为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通信连接可以是通过一些接口,单元或模块的间接耦合或通信连接,可以是电性或其它的形式。所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个单元上。可以根据实际的需要选择其中的部分或者全部单元来实现本实施例方案的目的。另外,在本发明各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。所述集成的单元如果以软件功能单元的形式实现并作为独立的产品销售或使用时,可以存储在一个计算机可读取存储介质中。基于这样的理解,本发明的技术方案本质上或者说对现有技术做出贡献的部分或者该技术方案的全部或部分可以以软件产品的形式体现出来,该计算机软件产品存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可为个人计算机、服务器或者网络设备等)执行本发明各个实施例所述方法的全部或部分步骤。而前述的存储介质包括:U盘、只读存储器(ROM,Read-OnlyMemory)、随机存取存储器(RAM,RandomAccessMemory)、移动硬盘、磁碟或者光盘等各种可以存储程序代码的介质。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 反向链路中用混合式自动重复请求进行软切换的闭环方法
- 用于单载波数字电视广播系统的重复pn1023序列回波抵消基准信号的制作方法
- 反向链路自动重复请求的制作方法
- 使隔行扫描产生的图象序列的图象重复频率加倍的方法
- 一种用于Ⅴ型腺病毒滴度检测的试剂盒及其应用的制作方法
- 一种新的多肽——含有CAG重复序列的异常人cDNA(B120)14.63和编码这种多肽的多核苷酸的制作方法
- 新的类似微型反向重复转座元件(mite)的转座元件和转录活化元件的制作方法
- 用于在音频帧序列中检测重复模式的方法及设备的制作方法
- 一种1型单纯疱疹病毒载体系统及其制备方法与应用的制作方法
- 一种基于人muc1重复序列与gm-csf融合表达的重组卡介苗疫苗的制作方法