本发明属于医药领域,涉及一种固体分散体及其制备,具体地一种改善吸收性能的固体分散体及其制备。
背景技术:
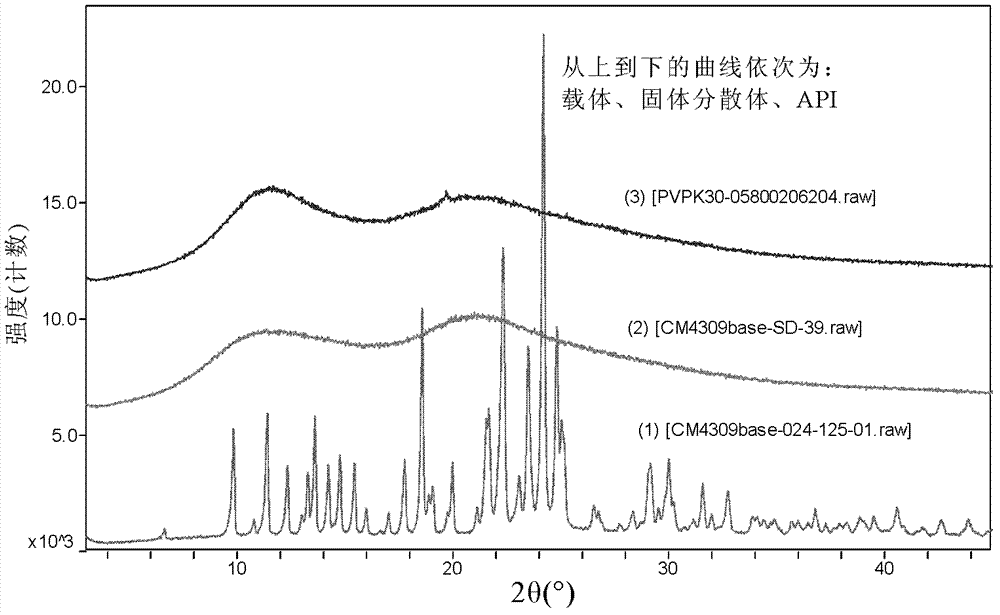
:4-(4-(3-(4-氯-3-(三氟甲基)苯基]酰脲)-3-氟-苯氧基)-2-(N-1’,1’,1’-三氘代甲基)吡啶酰胺,即式I化合物,所述化合物或其药学上可接受的盐对磷酸激酶(Kinase)例如raf激酶具有优异的抑制性,可用于治疗肿瘤及其相关疾病。该化合物为有色粉末,无臭无味。在二甲亚砜或二甲基甲酰胺中极易溶解,在四氢呋喃、甲醇中略溶,在丙酮、无水乙醇或冰醋酸中微溶,但是在水中几乎不溶。由于式I化合物水溶性极差,是难溶性药物,在生理体液中的溶解度和溶解速度较低,溶出度也较低,最终导致生物利用度较低,严重影响哺乳动物对式I化合物的吸收,不利于充分发挥该化合物的疾病治疗效果。固体分散体可以提高难溶性药物的生物利用度,但是现有的固体分散体,最高只能将药物的药时曲线下面积(AUC)提高2-6倍。例如,CN101632630A中,较普罗布考而言,普罗布考和PVPK30的固体分散体,其犬相对生物利用度也仅仅提高了5.8倍。因此,开发一种生物利用度显著提高的式I化合物的固体分散体,对解决哺乳动物对其活性成分吸收差的问题是非常有必要的。技术实现要素:本发明的一个目的是提供一种生物利用度显著提高的以式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物为活性成分的固体分散体。本发明的另一目的是提供本发明固体分散体的制备方法。本发明的再一目的是提供所述固体分散体的药物组合物。本发明第一方面提供了一种固体分散体,包括(A)作为活性成分的式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物,和式中,R为CD3或CH3;以及(B)亲水性高分子载体;其中,(A)和(B)的重量比为1∶1-1∶40。在另一优选例中,所述的亲水性高分子载体是聚维酮或含聚维酮的高分子载体。在另一优选例中,所述的亲水性高分子载体是聚维酮K30或含聚维酮K30的高分子载体,其中,聚维酮K30的含量为10-100wt%,按高分子载体的总重量计。在另一优选例中,所述活性成分为式I所示的化合物,或其药学上可接受的盐、水合物或溶剂合物:在另一优选例中,所述含聚维酮K30的高分子载体还包含选自下组的其他水溶性载体:聚乙二醇类、聚维酮类、纤维素类、表面活性剂类、或其组合;其中,所述聚乙二醇类选自下组:聚乙二醇4000、聚乙二醇6000、聚乙二醇8000或其组合;所述聚维酮类选自下组:聚维酮K25、聚维酮K90、聚维酮S630、或其组合;所述纤维素类选自下组:羟丙甲纤维素E5、羟丙甲纤维素E15、羟丙纤维素L、或其组合;所述表面活性剂类选自下组:泊洛沙姆407、泊洛沙姆188、或其组合。在另一优选例中,所述含聚维酮K30的高分子载体包含选自聚维酮K25、聚维酮K90、聚维酮S630、或其组合的聚维酮类载体。在另一优选例中,所述含聚维酮K30的高分子载体和其他水溶性载体的重量比例为1∶4-4∶1;较佳地为1∶2-4∶1;更佳地为1∶1-4∶1。在另一优选例中,所述(A)和(B)的重量比为1∶1-1∶20。在另一优选例中,所述(A)和(B)的重量比为1∶2-1∶9。在另一优选例中,所述固体分散体的X-射线粉末衍射图中,不具有所述固体分散体的活性成分的X-射线衍射特征峰。在另一优选例中,具有以下一个或多个特征:①所述固体分散体的药时曲线下面积是所述固体分散体的活性成分单独给药时的药时曲线下面积的6-20倍,较佳地7-15倍;②所述固体分散体在37℃、pH4.5的醋酸钠缓冲水溶液中10分钟时累积溶出度大于70%,较佳地为75-95%;③所述固体分散体在37℃、pH4.5的醋酸钠缓冲水溶液中120分钟时累积溶出度是所述固体分散体的活性成分累积溶出度的10-40倍,较佳地为15-30倍。在另一优选例中,所述的药时曲线下面积是在以下动物中测得的:大鼠、小鼠或犬。本发明第二方面提供了一种本发明第一方面所述固体分散体的制备方法,包括方法:(a)熔融法1,包括步骤:(a1)混合活性成分与亲水性高分子载体,从而得到一含活性成分的混合物;(a2)加热熔融步骤(a1)得到的含活性成分的混合物,从而得到熔融后的混合物;和(a3)冷却步骤(a2)得到的熔融后的混合物,从而得到本发明第一方面所述的固体分散体;或(b)熔融法2,包括步骤:(b1)加热熔融亲水性高分子载体,从而得到熔融后的载体;(b2)将步骤(b1)得到的熔融后的载体和活性成分混合后熔融,从而得到熔融后的混合物;和(b3)冷却步骤(b2)得到的熔融后的混合物,从而得到本发明第一方面所述的固体分散体;或(c)溶剂法,包括步骤:(c1)将活性成分与亲水性高分子载体共同溶解于溶剂中,从而得到一混合物;和(c2)除去步骤(c1)得到的混合物中的溶剂,从而得到本发明第一方面所述的固体分散体;其中,所述活性成分为式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物,式中,R为CD3或CH3,所述亲水性高分子载体为含聚维酮K30的亲水性高分子载体,所述溶剂选自下组:甲醇、乙醇、异丙醇、丙酮、二氯甲烷、四氢呋喃,或其组合。在另一优选例中,所述步骤(c2)包括步骤:用旋转蒸发仪除去步骤(c1)得到的混合物中的溶剂;或者用喷雾干燥器干燥除去步骤(c1)得到的混合物中的溶剂。在另一优选例中,所述干燥的工艺参数为:进风温度110℃~120℃,风机功率55%~65%,蠕动泵功率50%~55%。在另一优选例中,所述固体分散体进一步用至少一种另外的加工步骤处理,所述加工步骤为研磨、筛分、碾压、碾磨、筛选、混合或其组合。本发明第三方面提供了一种药物组合物,所述组合物包括:(1)本发明第一方面所述的固体分散体;和(2)药学上可接受的赋形剂。在另一优选例中,所述赋形剂包括:填充剂、稀释剂、甜味剂、矫味剂、抗氧化剂、着色剂、防腐剂、润滑剂、粘合剂、崩解剂、或其混合。在另一优选例中,所述组合物为胶囊剂、片剂、丸剂、散剂和颗粒剂。在另一优选例中,所述药物组合物,用于抑制磷酸激酶(如raf激酶)的药物组合物。在另一优选例中,所述的药物组合物用于治疗和预防癌症。本发明第四方面提供了一种本发明第三方面所述药物组合物的制备方法,将本发明第一方面所述的固体分散体和药学上可接受的赋形剂混合,从而制得所述的药物组合物。本发明第五方面提供了一种本发明第一方面所述的固体分散体的用途,用于制备抑制磷酸激酶(如raf激酶)的药物;或用于制备治疗肿瘤的药物。本发明第六方面提供了一种治疗方法,将(i)本发明第一方面所述的固体分散体或(ii)如本发明第三方面所述的药物组合物,施用于需要治疗的患者。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1显示了固体分散体1的XRPD图。图2显示了固体分散体2的XRPD图。图3显示了固体分散体3的XRPD图。图4显示了固体分散体4的XRPD图。图5显示了式I化合物、式I化合物与PVPK30的物理混合物和固体分散体1的溶出曲线。图6显示了式I化合物、式I化合物与PVPK30的物理混合物和固体分散体2的溶出曲线。图7显示了固体分散体10的XRPD图。图8显示了固体分散体10的溶出曲线。图9显示了固体分散体1经1、2、3个月加速稳定性试验后XRPD对比图,结果图9所示:从上往下依次是式I化合物、新制备的固体分散体1、固体分散体1放置一个月时、固体分散体1放置两个月时、固体分散体1放置三个月时的XRPD图。图10显示了固体分散体2经1、2、3个月加速稳定性试验后XRPD对比图,结果图10所示:从上往下依次是式I化合物、新制备的固体分散体2、固体分散体2放置一个月时、固体分散体2放置两个月时、固体分散体2放置三个月时的XRPD图。图11显示了固体分散体3经1、2、3个月加速稳定性试验后XRPD对比图,结果图11所示:从上往下依次是式I化合物、新制备的固体分散体3、固体分散体3放置一个月时、固体分散体3放置两个月时、固体分散体3放置三个月时的XRPD图。图12显示了固体分散体4经1、2、3个月加速稳定性试验后XRPD对比图,结果图12所示:从上往下依次是式I化合物、新制备的固体分散体4、固体分散体4放置一个月时、固体分散体4放置两个月时、固体分散体4放置三个月时的XRPD图。具体实施方式本发明人通过长期而深入的研究,意外地发现,一种包含(A)作为活性成分的式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物和(B)亲水性高分子载体(尤其是含聚维酮K30的高分子载体)的固体分散体,相对于活性成分而言,所述固体分散体的药时曲线下面积约为活性成分的6-20倍;所述固体分散体明显提高其活性成分的体外溶出度,显著改善哺乳动物对活性成分的吸收,从而有效地减小其使用剂量。此外,所述固体分散体还具有非常好的稳定性,极其适用于制备相关疾病的药物组合物。在此基础上,发明人完成了本发明。固体分散体固体分散体(soliddispersion)是指将活性成分高度分散于(固体)载体中形成的一种以固体形式存在的分散系统。活性成分如本文所用,术语“活性成分”是指式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物,式中,R为CD3或CH3;该术语还包括及式A化合物或其衍生物的各种晶型形式、水合物或溶剂合物。优选地,所述式A化合物指式I化合物或式II化合物、或其药学上可接受的盐。如本文所用,术语“药学上可接受的盐”指本发明化合物与酸或碱所形成的适合用作药物的盐。药学上可接受的盐包括无机盐和有机盐。一类优选的盐是本发明化合物与酸形成的盐。适合形成盐的酸包括但并不限于:盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸等无机酸,甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、苦味酸、甲磺酸、苯甲磺酸,苯磺酸等有机酸;以及天冬氨酸、谷氨酸等酸性氨基酸。优选地,所述药学上可接受的盐为对甲基苯磺酸盐。载体固体分散体所用的载体,最常用的有水溶性载体、难溶性载体、和肠溶性载体。本发明所述的“载体”为亲水性高分子载体,优选为聚维酮或含聚维酮的高分子载体,或者聚维酮K30或含聚维酮K30的高分子载体,其中,聚维酮K30的含量为10-100wt%,按高分子载体的总重量计。更优选地,本发明所用的含聚维酮K30的高分子载体是指聚维酮K30的重量含量超过载体总重量20%的载体;或聚维酮K30的重量含量超过载体总重量40%的载体;或聚维酮K30的重量含量超过载体总重量60%的载体。当然,还可以含有其他水溶性载体,例如包括(但不限于):聚乙二醇类、聚维酮类、纤维素类、表面活性剂类、甘露醇、或其组合。所述聚乙二醇类选自下组:聚乙二醇4000(PEG4000)、聚乙二醇6000(PEG6000)、聚乙二醇8000(PEG8000)或其组合;所述聚维酮类选自下组:聚维酮K90(PVPK90)、聚维酮K25(PVPK25)、聚维酮S630、或其组合;所述纤维素类选自下组:羟丙甲纤维素E5(HPMCE5)、羟丙甲纤维素E15(HPMCE15)、羟丙纤维素L(HPC-L)、或其组合;所述表面活性剂类选自下组:泊洛沙姆407(Poloxamer407)、泊洛沙姆188(Poloxamer188)、或其组合。组成本发明提供的固体分散体包括(A)作为活性成分的式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物,其中,式A化合物同上文中所述。和(B)亲水性高分子载体;其中,(A)和(B)的重量比为1∶1-1∶40。优选地,所述(A)和(B)的重量比为1∶20-1∶1,较佳地,所述(A)和(B)的重量比为1∶9-1∶2。制备方法本发明所述固体分散体可通过如下的方法制得,然而该方法的条件,例如载体、溶剂、各成分的量、制备温度、制备所需时间等不限于下面的解释。本发明所述固体分散体还可以任选将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便的制得,这样的组合可由本发明所属领域的技术人员容易的进行。具体地,所述制备方法可以是熔融法、溶剂法、溶剂-熔融法、或表面分散法。优选地,所述制备方法是熔融法、或溶剂法。熔融法通常的,所述熔融法是将(A)活性成分,和(B)亲水性高分子载体混合均匀,用水浴或油浴加热至熔融,然后冷却成固体,粉碎后得到粉末状的所述活性成分的固体分散体。或者,可将(B)亲水性高分子载体加热熔融后,再加入(A)活性成分,然后将熔融的混合物在剧烈搅拌下,迅速冷却成固体,粉碎后得到粉末状的所述活性成分的固体分散体。溶剂法通常的,所述溶剂法是将(A)活性成分,和(B)亲水性高分子载体共同溶解于溶剂中,混合物均匀后,除去溶液中的溶剂,可使活性成分和亲水性高分子载体同时析出,粉碎后即可得到粉末状的所述活性成分的固体分散体。所述溶剂法所使用的溶剂包括:甲醇、乙醇、异丙醇、丙酮、四氢呋喃、或其组合。结构鉴定固体分散体的结构鉴定方法包括差示扫描量热分析、X-射线粉末衍射等。分别对活性成分、载体和本发明所述方法制备的固体分散体,进行了X-射线粉末衍射(XRPD)检测,比较三者的XRPD谱图。活性成分的特征峰当制成本发明的固体分散体后,活性成分的X-射线衍射特征峰基本上或完全消失。换言之,本发明固体分散体的X-射线粉末衍射图中,不具有所述固体分散体的活性成分的X-射线衍射特征峰。式I化合物,其在7.24±0.1°、13.38±0.1°、14.53±0.1°、14.74±0.1°、15.61±0.1°、17.23±0.1°、18.57±0.1°、18.84±0.1°、19.81±0.1°、20.01±0.1°、23.42±0.1°、24.24±0.1°、25.85±0.1°、26.39±0.1°、27.43±0.1°有强衍射峰。式II化合物,其在7.20±0.1°、13.34±0.1°、14.49±0.1°、14.72±0.1°、15.55±0.1°、16.44±0.1°、17.17±0.1°、18.53±0.1°、18.81±0.1°、19.76±0.1°、19.97±0.1°、23.41±0.1°、23.75±0.1°、24.22±0.1°、24.75±0.1°、25.90±0.1°、26.35±0.1°、27.42±0.1°、31.54±0.1°有强衍射峰。载体的特征峰有些载体有强衍射峰,例如,PEG4000和PEG6000在19.20±0.1°、23.30±0.1°有强衍射峰。Poloxamer407在19.16±0.1°、23.33±0.1°有强衍射峰。有些载体没有强衍射峰,例如,PVPK30、HPMCE5和HPC-L均没有检测到强衍射峰。固体分散体的特征峰本发明制备的固体分散体的XRPD谱图中活性成分的强衍射峰消失,从而确定本发明所述的固体分散体制备成功,其中不是活性成分和载体的简单的物理混合物,而是以无定型或分子形态存在。药物组合物术语“本发明化合物”指式A化合物或其衍生物、或其药学上可接受的盐。术语“本发明固体分散体”指活性成分为式A化合物或其衍生物、或其药学上可接受的盐的固态分散体。由于本发明化合物具有优异的对磷酸激酶(Kinase)例如raf激酶的抑制活性,因此本发明固体分散体以及含有本发明固体分散体为主要成分的药物组合物可用于治疗、预防以及缓解由对磷酸激酶(Kinase)例如raf激酶介导的疾病。根据现有技术,本发明化合物可用于治疗以下疾病:癌症,心血管疾病,肥胖病,糖尿病等等。本发明的药物组合物包含安全有效量范围内的本发明固体分散体或其药理上可接受的盐及药理上可以接受的赋形剂。其中“安全有效量”指的是:本发明固体分散体的量足以明显改善病情,而不至于产生严重的副作用。通常,药物组合物含有1-2000mg本发明固体分散体/剂,更佳地,含有10-200mg本发明固体分散体/剂。较佳地,所述的“一剂”为一个胶囊或药片。“药学上可以接受的赋形剂”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。“相容性”在此指的是组合物中各组份能和本发明的固体分散体以及它们之间相互掺和,而不明显降低固体分散体的药效。药学上可以接受的赋形剂部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂(如吐温)、润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。本发明固体分散体或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)、和局部给药。用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在这些固体剂型中,本发明固体分散体与至少一种常规惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙,或与下述成分混合:(a)填料或增容剂,例如,淀粉、乳糖、蔗糖、葡萄糖、微晶纤维素、甘露醇和硅酸;(b)粘合剂,例如,羟丙甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯胶;(c)保湿剂,例如,甘油;(d)崩解剂,例如,琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合硅酸盐、交联羧甲基纤维素钠和碳酸钠;(e)缓溶剂,例如石蜡;(f)吸收加速剂,例如,季胺化合物;(g)润湿剂,例如鲸蜡醇、十二烷基硫酸钠和单硬脂酸甘油酯;(h)吸附剂,例如,高岭土;和(i)润滑剂,例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠,或其混合物。胶囊剂、片剂和丸剂中,剂型也可包含缓冲剂。固体剂型如片剂、糖丸、胶囊剂、丸剂和颗粒剂可采用包衣和壳材制备,如肠衣和其它本领域公知的材料。它们可包含不透明剂,并且,这种组合物中固体分散体的释放可以延迟的方式在消化道内的某一部分中释放。可采用的包埋组分的实例是聚合物质和蜡类物质。必要时,固体分散体也可与上述赋形剂中的一种或多种形成微胶囊形式。用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了固体分散体外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、娇味剂和香料。除了固体分散体外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。用于局部给药的本发明固体分散体的剂型包括软膏剂、散剂、贴剂、喷射剂和吸入剂。固体分散体在无菌条件下与生理上可接受的载体及任何防腐剂、缓冲剂,或必要时可能需要的推进剂一起混合。本发明固体分散体可以单独给药,或者与其他药学上可接受的化合物联合给药。使用药物组合物时,是将安全有效量的本发明固体分散体适用于需要治疗的哺乳动物(如人),其中施用时剂量为药学上认为的有效给药剂量,对于60kg体重的人而言,日给药剂量通常为1~2000mg,优选20~500mg。当然,具体剂量还应考虑给药途径、病人健康状况等因素,这些都是熟练医师技能范围之内的。本发明的主要优点是:(1)提供了一种以式A化合物或其衍生物、或其药学上可接受的盐、水合物或溶剂合物为活性成分的固体分散体,所述固体分散体显著改善了活性成分的体外溶出度,非常有利于哺乳动物对该活性成分的吸收。而且相对于其活性成分化合物本身而言,所述固体分散体具有显著优异的体内药代动力学参数(尤其是药时曲线下面积),且非常稳定。(2)提供了一种所述固体分散体的制备方法。下面结合具体实施,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。如本文所用“API”是指活性成分。实施例固体分散体的溶出度测试方法按照《中华人民共和国药典》2010年版二部附录XC溶出度测定法第二法(桨法)进行检测。条件如下:溶出介质:0.1%十二烷基硫酸钠pH4.5醋酸-醋酸钠缓冲液900mL/溶出杯;温度:37±0.5℃;转速:100转/分钟。具体地,将样品分为三组,每组样品分别配置3份。其中,第1组样品是活性成分;第2组样品是只是将活性成分与载体通过简单的物理方法(如搅拌)混合后的样品。第三组样品是按照各个实施例中制备方法得到的固体分散体。具体如下表。使用所述溶出介质,按照常规方法分别检测以上3组样品的溶出度,每组检测3份样品,取其平均值。比较3组的测试结果。实施例1固体分散体1式I化合物与PVPK30,重量比例为1∶9,溶剂(无水乙醇)法称取1重量份的式I化合物和9重量份的PVPK30置同一烧瓶中,烧瓶置80℃油浴中,加入无水乙醇至烧瓶中,密塞在冷凝水回流条件下搅拌,直至化合物和PVPK30均溶清,再搅拌30分钟。将溶清的液体用旋转蒸发器在80℃时快速蒸干,得到固体。将固体在50℃真空干燥箱内干燥24小时,研细,即得固体分散体1。其XRPD测试结果如图1所示,固体分散体1中,式I化合物的衍射峰(特征峰)全部消失,以无定型或分子形态存在。实施例2固体分散体2式I化合物与PVPK30,重量比例为1∶4,溶剂(无水乙醇)法制备方法同实施例1,不同点在于式I化合物与PVPK30的重量比例为1∶4。其XRPD测试结果如图2所示,固体分散体2中,式I化合物的衍射峰(特征峰)全部消失,以无定型或分子形态存在。实施例3固体分散体3式I化合物与PVPK30,重量比例为1∶3,溶剂(无水乙醇)法制备方法同实施例1,不同点在于式I化合物与PVPK30的重量比例为1∶3。其XRPD测试结果如图3所示,固体分散体3中,式I化合物的衍射峰(特征峰)全部消失,以无定型或分子形态存在。实施例4固体分散体4式I化合物与PVPK30,重量比例为1∶2,溶剂(无水乙醇)法制备方法同实施例1,不同点在于式I化合物与PVPK30的重量比例为1∶2。其XRPD测试结果如图4所示,固体分散体4中,式I化合物的衍射峰(特征峰)全部消失,以无定型或分子形态存在。实施例5-9固体分散体5-9制备方法同实施例1,具体条件见表1。表1实施例活性成分/载体(重量比)溶剂5式I化合物/PVPK90(1∶4)无水乙醇6式I化合物/PVPK25(1∶4)无水乙醇7式I化合物/PVPS630(1∶4)无水乙醇8式I化合物/HPMCE5(1∶5)无水乙醇9式I化合物/HPC-L(1∶5)丙酮实施例10固体分散体1的溶出度测试参考中国药典2010版二部附录XC溶出度测试法,量取0.1%十二烷基硫酸钠pH4.5醋酸-醋酸钠缓冲液900mL,置于1000mL溶出杯中,温度保持37±0.5℃。分别加入式I化合物、式I化合物和PVPK30的物理混合物(式I化合物和PVPK30的重量分别为50mg和450mg)、固体分散体1(500mg),桨速为100rpm,分别于2min、5min、10min、15min、20min、30min、45min、60min、120min取样5mL,用0.45μm微孔滤膜过滤,取澄清滤液稀释后测定药物浓度,计算三者的溶出度。结果如图5所示,式I化合物在10min时的溶出度<2%,式I化合物和PVPK30的物理混合物的溶出度<5%,固体分散体1的溶出度为78%,以PVPK30为基质的固体分散体极大地改善了式I化合物的溶出度。实施例11固体分散体2的溶出度测试溶出度测试方法同实施例10。测试结果如图6所示,30min时,式I化合物的溶出度<2%,式I化合物和PVPK30的物理混合物的溶出度<5%,固体分散体2(载体为PVPK30)的溶出度为78%。120min时,式I化合物的溶出≤5%,式I化合物与载体的物理混合物的溶出略好于式I化合物的,但≤10%,式I化合物与载体形成的固体分散体的溶出约80%。结果表明:固体分散体的溶出度要明显好于式I化合物及式I化合物与载体的物理混合物。此外,PVPK30为载体的固体分散体均极大的改善了式I化合物的溶出度,显著优于采用其他常用的载体(如PEG6000、羟丙甲纤维素E5),也优于PVPK90和K25(30分钟时溶出度65-70%)。实施例12固体分散体2的体内药代研究选取固体分散体2与对应的API(即式I化合物)进行体内药代试验。8只雄性6周龄Wistar大鼠,体重170g左右,分为2组,每组4只。动物饲养于IVC动物房内,12小时明暗交替。以硬木刨花作为垫料。动物给予饲料和经灭菌的自来水,自由饮食。在给药前禁食16h,给药后2h恢复给食。禁食过夜后,灌胃给予4mg/kg固体分散体1或者对应的API的1%CMC悬液,给药容量为10ml/kg。于给药后0.5、1.0、2.0、4.0、6.0、8.0、24、48和72h经大鼠眼球后静脉丛取静脉血0.2ml,置肝素化试管中,3500rpm离心10min,分离得到血浆样品,-20℃保存。血浆样品用经过验证的LC-MS/MS分析方法定量检测API的含量。药代动力学参数将基于每只大鼠在不同时间点的血药浓度进行计算。参数参见表2。结果如表2所示:式I化合物与PVPK30按1∶4重量比制得的固体分散体2体内药代的AUC0-∞约为式I化合物的9.03倍。表2实施例13固体分散体10式II化合物与PVPK30,重量比例为1∶4,溶剂(无水乙醇)法制备方法同实施例1,不同点在于式II化合物代替了式I化合物,式II化合物与PVPK30的重量比例为1∶4。其XRPD测试结果如图7所示,固体分散体10中,式II化合物的衍射峰(特征峰)全部消失,以无定型或分子形态存在。实施例14固体分散体10的溶出度测试溶出度测试方法同实施例10。具体地,将样品配置2份(分别为sample1和sample2)。按照上述方法测试各自的溶出度,并计算平均值(即为average)。测试结果如图8所示,30min时,固体分散体10的溶出度约为72.5%,120min时,固体分散体10的溶出约为94%。可见,式II化合物与载体PVPK30形成的固体分散体10的溶出度很好。实施例15固体分散体10的体内药代研究选取固体分散体10与对应的API(即式II化合物)进行体内药代试验。测试方法同实施例12。结果如表3所示:式II化合物与PVPK30按1∶4重量比例制备的固体分散体10体内药代的AUClast约为式II化合物的13.5倍。表3实施例16固体分散体1加速稳定性试验选取固体分散体1进行加速稳定性试验(40℃,75%RH),1、2、3个月时均取出进行X-射线粉末衍射。XRPD结果如图9所示,表明固体分散体1经1、2、3个月加速稳定性试验后,非常稳定(无API的X-衍射特征峰,且曲线基本相同),均没有晶型生成(即原来的无定型或分子状态没有改变),仍然是以无定形或分子状态存在的固体分散体。结果图9所示:从上往下依次是式I化合物、新制备的固体分散体1、固体分散体1放置一个月时、固体分散体1放置两个月时、固体分散体1放置三个月时的XRPD图。实施例17固体分散体2加速稳定性试验选取固体分散体2进行加速稳定性试验(40℃,75%RH),1、2、3个月时均取出进行X-射线粉末衍射。XRPD结果如图10所示,表明固体分散体2经1、2、3个月加速稳定性试验后,非常稳定(无API的X-衍射特征峰,且曲线基本相同),均没有晶型生成(即原来的无定型或分子状态没有改变),仍然是以无定形或分子状态存在的固体分散体。结果图10所示:从上往下依次是式I化合物、新制备的固体分散体2、固体分散体2放置一个月时、固体分散体2放置两个月时、固体分散体2放置三个月时的XRPD图。实施例18固体分散体3加速稳定性试验选取固体分散体3进行加速稳定性试验(40℃,75%RH),1、2、3个月时均取出进行X-射线粉末衍射。XRPD结果如图11所示,表明固体分散体3经1、2、3个月加速稳定性试验后,非常稳定(无API的X-衍射特征峰,且曲线基本相同),均没有晶型生成(即原来的无定型或分子状态没有改变),仍然是以无定形或分子状态存在的固体分散体。结果图11所示:从上往下依次是式I化合物、新制备的固体分散体3、固体分散体3放置一个月时、固体分散体3放置两个月时、固体分散体3放置三个月时的XRPD图。实施例19固体分散体4加速稳定性试验选取固体分散体4进行加速稳定性试验(40℃,75%RH),1、2、3个月时均取出进行X-射线粉末衍射。XRPD结果如图12所示,表明固体分散体4经1、2、3个月加速稳定性试验后,非常稳定(无API的X-衍射特征峰,且曲线基本相同),均没有晶型生成(即原来的无定型或分子状态没有改变),仍然是以无定形或分子状态存在的固体分散体。结果图12所示:从上往下依次是式I化合物、新制备的固体分散体4、固体分散体4放置一个月时、固体分散体4放置两个月时、固体分散体4放置三个月时的XRPD图。实施例20-24固体分散体11-15活性成分:式I化合物载体:PVPK30和其他载体的混合载体制备方法同实施例1,不同点在于混合载体采用如表4所示的条件。表4其XRPD测试结果表明,固体分散体中,式I化合物的衍射峰(特征峰)全部消失,以无定型形态存在。固体分散体11-15的体内药代试验(操作同前)表明:固体分散体的体内药代的AUClast约为API的7-15倍。可见,本发明制备的包含活性成分和含PVPK30的载体的固体分散体非常稳定;且较活性成分而言,本发明的固体分散体具有显著提高的溶出度,尤其是本发明所述固体分散体的药时曲线下面积是其活性成分单独给药时的药时曲线下面积的6-20倍,较佳地7-15倍,更佳地为9-14倍。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页1 2 3