一种无定型依帕列净的固体分散体及其制备方法与流程
本发明属于药物制剂领域,具体涉及一种无定型依帕列净的固体分散体及其制备方法。本发明还涉及一种药用组合物,该组合物包含无定形态依帕列净的固体分散体和至少一种的药学上可接受的辅料,用于治疗糖尿病。
背景技术:
依帕列净(Emagliflozin),商品名Jardiance,化学名称为:(1S)-1,5-脱水-1-C-[4-氯-3-[[4-[[(3S)-四氢-3-呋喃基]氧基]苯基]甲基]苯基]-D-葡萄糖醇,其结构如式(I)所示:
式I
依帕列净是由德国勃林格英格翰公司和美国礼来公司联合研发的II型糖尿病治疗用药。依帕列净于2014年5月22日在欧盟获批上市,并于2014年8月1日被美国FDA批准上市。
据预测,未来10-20年糖尿病患病人数将居所有疾病之首,到2020年中国糖尿病患者人数将高达1.5亿,为全球糖尿病患者最多的国家。全球糖尿病市场总销售额已超过350亿美元,中国糖尿病市场规模已达150亿。依帕列净是全球第3个上市的糖尿病治疗新靶点钠-葡萄糖协同转运蛋2(SGLT-2)抑制剂,有利于2 型糖尿病患者的血糖调控,并且不会造成低血糖,并有一定的减肥效果,为病人提供了更好的选择。
依帕列净存在多种晶型。中国专利CN101155794公开了一种依帕列净晶型。中国专利CN104788438页公开了一种依帕列净的晶型Form B。印度专利IN 2013MU01985公开了一种无定型态的依帕列净以及一种含无定型态依帕列净和一种高分子材料的固体分散体。
药物的固体形态直接影响原料药的溶解速率、制剂的溶出度和生物利用度,为了提高药物的生物利用度,降低用量、降低毒副作用,通常会开发药物的新的固体形态,因此,开发该药物溶解性更好、生物利用度更高的固体形式就显得很有必要。
药物的固体形态除晶态外,还有无定型状态,药物的无定型状态作为固体物质的一种特殊形态,在药物制备中有着重要的用途。一般由于晶态物质分子的有序和周期性排列,降低了分子间相互作用的能量,能量较低,而无定型态的分子处于高度无序状态,物质的表面自由能更大,固体物质中的分子较晶态固体物质中的分子有更高的能量,更容易分散,增加其溶出度,提高药物的生物利用度。无定型态药物不仅可以广泛应用于药物制剂中,而且可以通过多种技术手段和方法提高无定型态药物的稳定性,使之成为具有优良品质的药物。
印度专利IN 2013MU01985公开了一种无定型态的依帕列净以及一种含无定型态依帕列净和一种高分子材料的固体分散体。该固体分散体中只含有一种高分子药用辅料。由于单一的高分子药用辅料对药物的分散作用并不是非常理想,通常所得固体分散体的化学稳定性和物理稳定性都会变差。所以要得到无定型药物分子并使之稳定在无定型状态往往需要比较大的药用辅料的量。药物制剂须用到多种药用辅料,且辅料总量也有一定限制,如某种单一辅料的用量较大时,也会对药物制剂配方的开发带来一定困难。另外,对于只加入一种高分子辅料的固体分散体的分离得不到易分离的固体。如果使用两种或两种以上的药用辅料,往往可以改善固体分散体的物理状态,使之便于分离,更有利于大规模的工业化生产。
由于现有的无定形态依帕列净的不足和无定型药物活性成分在药物制剂方面的良好的应用前景,寻找新的无定型依帕列净及其制备方法就显得十分必要。
技术实现要素:
本发明的目的是提供一种依帕列净与药用辅料的固体分散体及其制备方法,得到稳定性及分散性良好的无定型态的依帕列净与药用辅料的固体分散体,增加了依帕列净的溶出度,该制备方法不受干燥过程的限制,也不受溶剂种类和溶剂量的限制,操作简便,成本低廉,易于实现,可实现工业化生产。
为了达到上述目的,本发明的技术方案如下:
一种依帕列净与药用辅料的固体分散体,该固体分散体包含依帕列净与两种或两种以上的药用辅料,依帕列净与全部药用辅料的重量比为1:0.1~100,其中,所述固体分散体中的依帕列净为无定型态,所述固体分散体的X-射线粉末衍射光谱中,扣除药用辅料的背景峰后无依帕列净的晶体的特征峰。
进一步,所述药用辅料中的至少一种选自稀释剂、润滑剂、粘合剂、崩解剂、表面活性剂、成膜材料、包衣材料和胶囊材料中的至少一种。
优选地,所述药用辅料中的至少一种选自羟丙甲基纤维素、羟丙基纤维素、聚维酮、聚乙二醇、乙基纤维素、微晶纤维素、脂质体、甲基丙烯酸共聚物、聚醋酸乙烯、羧甲基乙基纤维素、羧甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素醋酸酯琥珀酸酯、聚丙烯酸树脂、聚羧乙烯、藻酸盐、卡拉胶、羧基乙酸内酯、树胶、聚乙烯醇、预胶化淀粉、交联淀粉、羧甲基淀粉钠、糊精、聚环氧乙烷、壳聚糖、几丁聚糖、胶原蛋白和环糊精中的至少一种。
本发明的依帕列净与药用辅料的固体分散体的制备方法,包括如下步骤:
1)将依帕列净和两种或两种以上的药用辅料混合,加热至药用辅料熔融;其中,依帕列净与全部药用辅料的重量比为1:0.1~100;
2)混合均匀后冷却,将混合物粉碎,得到无定型态的依帕列净与药用辅料的固体分散体。
进一步,所述药用辅料中的至少一种选自稀释剂、润滑剂、粘合剂、崩解剂、表面活性剂、成膜材料、包衣材料和胶囊材料中的至少一种。
优选地,步骤1)中所述的药用辅料中的至少一种选自羟丙甲基纤维素、羟丙基纤维素、聚维酮、聚乙二醇、乙基纤维素、微晶纤维素、脂质体、甲基丙烯酸共聚物、聚醋酸乙烯、羧甲基乙基纤维素、羧甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素醋酸酯琥珀酸酯、聚丙烯酸树脂、聚羧乙烯、藻酸盐、卡拉胶、羧基乙酸内酯、树胶、聚乙烯醇、预胶化淀粉、交联淀粉、羧甲基淀粉钠、糊精、聚环氧乙烷、壳聚糖、几丁聚糖、胶原蛋白和环糊精中的至少一种。
本发明提供另一种依帕列净与药用辅料的固体分散体的制备方法,包括如下步骤:
1) 将依帕列净和两种或两种以上的药用辅料在溶剂中混合,混合温度为-50~150℃,形成含依帕列净和药用辅料的溶液或悬浮液,其中,依帕列净与溶剂的重量比为0.001~100:1,依帕列净与全部药用辅料的重量比为1:0.1~100;
2) 除去步骤1)得到的溶液或悬浮液中的溶剂,得到无定型态的依帕列净与药用辅料的固体分散体。
进一步,所述药用辅料中的至少一种选自稀释剂、润滑剂、粘合剂、崩解剂、表面活性剂、成膜材料、包衣材料和胶囊材料中的至少一种。
优选地,步骤1)中所述的药用辅料中的至少一种选自羟丙甲基纤维素、羟丙基纤维素、聚维酮、聚乙二醇、乙基纤维素、脂质体、甲基丙烯酸共聚物、聚醋酸乙烯、羧甲基乙基纤维素、羧甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素醋酸酯琥珀酸酯、聚丙烯酸树脂、聚羧乙烯、藻酸盐、卡拉胶、羧基乙酸内酯、树胶、聚乙烯醇、预胶化淀粉、交联淀粉、羧甲基淀粉钠、糊精、聚环氧乙烷、壳聚糖、几丁聚糖、胶原蛋白和环糊精中的至少一种。
又,步骤1)所述溶剂选自含12个以下碳原子的醇类、酚类、醚类、卤代烃、酮类、醛类、腈类、酰胺、砜、亚砜、羧酸和水中的至少一种,步骤2)除去溶剂的方法包括:蒸发、真空蒸发、喷雾干燥、冷冻干燥、热熔挤出、过滤、离心或搅拌薄膜干燥。
本发明还提供了一种药用组合物,所述药用组合物含有无定型依帕列净和两种或两种以上的药学上可接受的辅料,所述药用辅料中的至少一种选自稀释剂、润滑剂、粘合剂、崩解剂、表面活性剂、成膜材料、包衣材料和胶囊材料中的至少一种。
进一步,上述药用组合物中的药用辅料中的至少一种选自羟丙甲基纤维素、羟丙基纤维素、聚维酮、聚乙二醇、乙基纤维素、微晶纤维素、脂质体、甲基丙烯酸共聚物、聚醋酸乙烯、羧甲基乙基纤维素、羧甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素邻苯二甲酸酯、羟丙甲基纤维素醋酸酯琥珀酸酯、聚丙烯酸树脂、聚羧乙烯、藻酸盐、卡拉胶、羧基乙酸内酯、树胶、聚乙烯醇、预胶化淀粉、交联淀粉、羧甲基淀粉钠、糊精、聚环氧乙烷、壳聚糖、几丁聚糖、胶原蛋白和环糊精中的至少一种。
本发明还提供一种药用组合物,该组合物含有无定型依帕列净固体分散体和至少一种药学上可接受的辅料。该组合物中的固体分散体包含无定型依帕列净和至少两种药学上可接受的辅料。
进一步,该组合物用于制备抗糖尿病药物。
本发明的依帕列净与药用辅料的固体分散体,使用Cu-Kα辐射,以度2θ表示的X-射线粉末衍射光谱中扣除药用辅料的背景峰无依帕列净结晶态的特征峰,表明依帕列净为无定型状态。现有技术中一般使用依帕列净的结晶态,未见其无定型态的报道。一般由于晶态物质分子的有序和周期性排列,降低了分子间相互作用的能量,能量较低,而本发明的依帕列净为无定型态,分子处于高度无序状态,物质的表面自由能更大,固体物质中的分子较晶态固体物质中的分子有更高的能量,更容易分散,增加其溶出度,提高依帕列净的生物利用度。
本发明将依帕列净和药用辅料混合均匀后,使用“固体分散剂”法,通过药用辅料的多聚体网状结构将药物分子阻隔,抑制结晶的发生,使其保持分散和无定型状态。本发明中的药用辅料选用两种或两种以上的药用辅料。与单一辅料相比,多种药用辅料相互配伍,可以更好地发挥分散、阻隔药物分子和抑制结晶的作用。例如,针对依帕列净具有多个羟基的这一结构特点,本发明在药用辅料中引入多羟基的醇类,药物分子和醇类的羟基之间易形成氢键,产生较强的相互作用,可增强药物在辅料中的分散度,并能更好地抑制药物分子的结晶。此外,多种药用辅料也可以在药物制剂中发挥不同的作用,有利于药物制剂的开发。本发明采用应用广泛、价格低廉、溶解性好的药用辅料,这些药用辅料与依帕列净混合,配合蒸发、喷雾干燥、冷冻干燥和热熔挤出等技术可以得到依帕列净的无定型形式,增加本发明依帕列净的固体分散体中的依帕列净的无定型态的稳定性。所得固体物理状态良好,易于分离。
本发明选用在药学上应用广泛的、价格低廉的辅料,得到依帕列净与药用辅料的固体分散体,易于开发制剂配方,本发明的制备方法不受干燥过程的限制,也不受溶剂种类和溶剂量的限制,操作简便,便于分离,成本低廉,易于实现,可实现工业化生产。
与现有技术相比,本发明的有益效果是:
1) 本发明制备的无定型依帕列净与两种或两种以上的药用辅料的固体分散体具有高度分散性及稳定性,各种药用辅料可在药物制剂中附会不同的作用,有利于制剂配方的开发。在制成固体制剂后,经过崩解可使药物粒子的分散程度更好,分散及溶出速度更快,有利于药物的吸收。因此,无定型状态药物的溶出度明显增加,更有利于机体对药物的吸收,提高药物的生物利用度,使药物能够更好地发挥临床疾病治疗作用。
2) 本发明无定型状态的依帕列净与药用辅料的固体分散体的制备方法不受干燥过程的限制,也不受溶剂种类和溶剂量的限制,操作简便,便于分离,成本低廉,易于实现,可实现工业化生产。
3) 本发明制备的无定型状态的依帕列净与药用辅料的固体分散体在高温、高湿条件下,有关物质无显著改变,无依帕列净结晶析出;在加速试验条件下(40±2℃,湿度75%±5%),有关物质无显著改变,无依帕列净结晶析出,本发明的无定型状态的依帕列净与药用辅料的固体分散体能保持良好的物理稳定性和化学稳定性,将会有广阔的应用前景。
附图说明
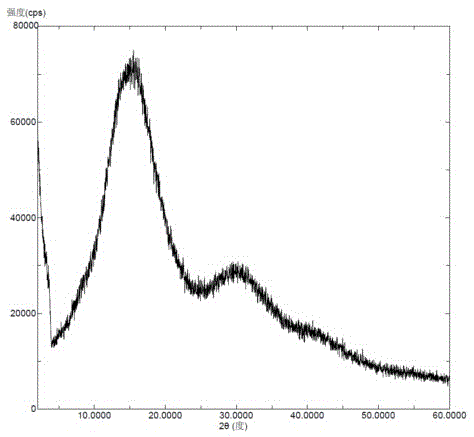
图1为本发明实施例1的无定型依帕列净和羟丙基纤维素SSL及聚维酮K30的固体分散体的X-射线粉末衍射图。
图2为本发明实施例12的无定型依帕列净和聚乙二醇4000及聚丙烯酸树脂L100的固体分散体的X-射线粉末衍射图。
具体实施方式
以下结合具体实施例对本发明作进一步说明,但本发明的保护范围不受以下实施例的限制。
本发明所述的X-射线粉末衍射图在Ultima IV X-射线衍射仪上采集。本发明所述的X-射线粉末衍射的方法参数如下:
X-射线粉末参数:Cu-Kα
Kα():1.5418
电压:40千伏
电流:40毫安
发散狭缝:自动
扫描模式:连续
扫描范围:自2.0至60.0度
取样步长:0.0200度
扫描速率:60度/分钟
实施例1
将依帕列净(50毫克)、羟丙基纤维素SSL(50毫克)和聚维酮K30(50毫克)加入到甲醇(800微升)中,加热到60℃搅拌溶清,真空蒸发除去溶剂,得到白色粉末状固体,即无定型依帕列净与羟丙基纤维素SSL及聚维酮K30的固体分散体。该固体分散体的X-射线粉末衍射图如图1所示,X-射线粉末衍射图中扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例2
将依帕列净(50毫克)、聚丙烯酸树脂Eudragit L100(50毫克)和聚乙二醇4000(200毫克)溶于乙醇(600微升)和水(600微升)中,在-40℃下搅拌混合均匀,真空蒸发除去溶剂,得到白色粉末状固体,即无定型依帕列净与聚丙烯酸树脂Eudragit L100及聚乙二醇4000的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例3
将依帕列净(2克)、糊精(2克)和聚乙二醇8000(10克)加入水(300毫升)中,加热到60℃搅拌溶清。将上述溶液用JISL微型喷雾干燥机LSD-48干燥,维持进口温度60℃、出口温度50℃,收集出口物料,得到白色粉末状固体,进一步真空干燥得到无定型依帕列净与糊精及聚乙二醇8000的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例4
将依帕列净(1克)、β-环糊精(1克)和羟丙甲基纤维素E50(0.2克)加到水(10毫升)中,加热到40℃搅拌溶清。将上述溶液冷冻干燥,得到白色固体,即无定型依帕列净与β-环糊精及羟丙甲基纤维素E50的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例5
将依帕列净(5克)、聚维酮K30(10克)和聚乙二醇8000(50克)加热到熔融,搅拌下迅速冷却到室温,得到白色固体。将上述固体粉碎,得到白色粉末状固体,即无定型依帕列净与聚维酮K30及聚乙二醇8000的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例6
将依帕列净(1克)、乙醇(0.1克)、聚维酮K90(1克)和聚乙二醇10000(20克)加热到240℃,混合均匀,迅速冷却到室温,得到白色固体。将上述固体粉碎,得到白色粉末状固体,即无定型依帕列净与聚维酮K90及聚乙二醇10000的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例7
将依帕列净(1克)、羟丙基纤维素SSL (2克)、四氢呋喃(10克)、乙醇(20克)和脂质体(4克)的混合物加热到60℃,搅拌,混合均匀,真空蒸发除去溶剂,冷却到室温得到白色粉末状固体,即无定型依帕列净与羟丙基纤维素SSL及脂质体的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例8
将依帕列净(1克)、甲醇(20克)、聚丙烯酸树脂Eudragit L100(2克)和甲基丙烯酸共聚物A型(4克)的混合物加热到50℃,搅拌,溶清,迅速冷却到-30℃,得到白色粉末状固体,即无定型依帕列净与聚丙烯酸树脂Eudragit L100及甲基丙烯酸共聚物A型的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例9
将依帕列净(1克)、甲醇(20克)、预胶化淀粉(1克)和乙基纤维素(2克)的混合物加热到30℃,搅拌,混合均匀,真空蒸发除去溶剂,冷却到室温得到白色粉末状固体,即无定型依帕列净与预胶化及乙基纤维素的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例10
将依帕列净(1克)、甲醇(20克)、预胶化淀粉(2克)和羟丙基纤维素SSL(4克)的混合物加热到30℃,搅拌溶清,真空蒸发除去溶剂,冷却到室温得到白色粉末状固体,即无定型依帕列净与预胶化淀粉及羟丙基纤维素SSL的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例11
将依帕列净(1克)、甲醇(20克)、水(10克)、微晶纤维素(1克)和聚醋酸乙烯(4克)的混合物加热到30℃,搅拌溶清,真空蒸发除去溶剂,冷却到室温得到白色粉末状固体,即无定型依帕列净与微晶纤维素及聚醋酸乙烯的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例12
将依帕列净(50毫克)、聚乙二醇4000(100毫克)和聚丙烯酸树脂Eudragit L100(100毫克)加入到甲醇(750微升),室温下搅拌溶清,真空蒸发除去溶剂,得到白色粉末状固体,即无定型依帕列净与聚乙二醇4000及聚丙烯酸树脂Eudragit L100的固体分散体。该固体分散体的X-射线粉末衍射图如图2所示,X-射线粉末衍射图中扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例13
将依帕列净(50毫克)羧甲基纤维素邻苯二甲酸酯Agucoat CPD(2毫克)和聚丙烯酸树脂Eudragit S100(3毫克)加入到甲醇(4毫升)和乙酸乙酯(1毫升),在-30℃下搅拌溶清。将上述溶液在旋转蒸发其上浓缩除去溶剂,冷却至室温,得到白色粉末状固体,即无定型依帕列净与羧甲基纤维素邻苯二甲酸酯Agucoat CPD及聚丙烯酸树脂Eudragit S100的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例14
将依帕列净(50毫克)、糊精(50毫克)和聚羧乙烯Carbomer 940(50毫克)加入到甲醇(4毫升)和四氢呋喃(1毫升),在-30℃下搅拌混合均匀。将上述溶液在旋转蒸发其上浓缩除去溶剂,冷却至室温,得到白色粉末状固体,即无定型依帕列净与糊精及聚羧乙烯Carbomer 940的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例15
将依帕列净(50毫克)、β-环糊精(100毫克)和预胶化淀粉Pharma-Gel(100毫克)加入到甲醇(4毫升)和水(1毫升),室温下混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与β-环糊精及预胶化淀粉Pharma-Gel的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例16
将依帕列净(50毫克)、β-环糊精(100毫克)和高支链交联淀粉(50毫克)加入到甲醇(4毫升)和水(1毫升),室温下搅拌溶清,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与β-环糊精(100毫克)及高支链交联淀粉的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例17
将依帕列净(50毫克)、微晶纤维素(100毫克)和羧甲基纤维素钠SCMC(500毫克)加入到二甲基亚砜(5毫升),室温下搅拌溶清,真空蒸发除去溶剂,得到白色粉末状固体,进一步真空干燥,即无定型依帕列净与微晶纤维素及羧甲基纤维素钠SCMC的固体分散体,该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例18
将依帕列净(50毫克)、聚乙二醇4000(100毫克)和几丁聚糖(400毫克)加入到乙醇(5毫升),室温下搅拌溶清,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与聚乙二醇4000及几丁聚糖的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例19
将依帕列净(50毫克)、微晶纤维素(50毫克)和羧甲基淀粉钠Explotab(500毫克)加入到乙醇(5毫升),室温下搅拌混合均匀,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与D-微晶纤维素及羧甲基淀粉钠Explotab的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例20
将依帕列净(50毫克)、聚维酮K90(100毫克)和藻酸盐E401(100毫克)加入到乙醇(5毫升),室温下搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与聚维酮K90及藻酸盐E401的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例21
将依帕列净(50毫克)、微晶纤维素(100毫克)和羧甲基纤维素邻苯二甲酸酯Agucoat CPD(1克)悬浮于甲醇(30毫升),加热到50℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去大部分溶剂,过滤,干燥,得到白色粉末状固体,即无定型依帕列净与微晶纤维素及羧甲基纤维素邻苯二甲酸酯Agucoat CPD的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例22
将依帕列净(50毫克)、树胶Galactosol(100毫克)和卡拉胶E407(100毫克)悬浮于甲醇(30毫升),加热到50℃搅拌混合均匀,将上述溶液在旋转蒸发器中缓慢浓缩除去大部分溶剂,过滤,干燥,得到白色粉末状固体,即无定型依帕列净与树胶Galactosol及卡拉胶E407的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例23
将依帕列净(50毫克)、微晶纤维素(100毫克)和壳聚糖(200毫克)悬浮于甲醇(50毫升),加热到50℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去大部分溶剂,过滤,干燥,得到白色粉末状固体,即无定型依帕列净与微晶纤维素及壳聚糖的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例24
将依帕列净(300毫克)、脂质体(300毫克)和聚丙烯酸树脂 Eudragit E100(300毫克)溶于乙醇(600微升)、四氢呋喃(900微升)和N,N-二甲基甲酰胺(600微升)中,加热到50℃搅拌溶清,将上述溶液降温到-30℃,析出白色粉末状固体,过滤,干燥,得到无定型依帕列净与脂质体及聚丙烯酸树脂 Eudragit E100的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例25
将依帕列净(30毫克)、聚乙二醇8000(30毫克)和胶原蛋白Peptan(200毫克)溶于乙醇(600微升)和乙腈(600微升)中,加热到50℃搅拌溶清。将上述溶液在旋转蒸发器中缓慢浓缩除去大部分溶剂,析出白色粉末状固体,过滤,干燥,得到无定型依帕列净与聚乙二醇8000及胶原蛋白Peptan的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例26
将依帕列净(30毫克)、聚乙二醇8000(30毫克)和树胶Galactosol(150毫克)溶于甲醇(900微升)加热到50℃搅拌溶清。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即得到无定型依帕列净与聚乙二醇8000及树胶Galactosol的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例27
将依帕列净(30毫克)、几丁聚糖(30毫克)和羟丙甲基纤维素邻苯二甲酸酯HPMCP(30毫克)加入到乙醇(750微升)和水(750微升),加热到80℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与几丁聚糖及羟丙甲基纤维素邻苯二甲酸酯HPMCP的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例28
将依帕列净(30毫克)、微晶纤维素(30毫克)和羧基乙酸内酯(300毫克)加入到乙醇(750微升)和水(750微升),加热到80℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与微晶纤维素及羧基乙酸内酯的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例29
将依帕列净(30毫克)、β-环糊精(60毫克)和糊精Maltrin M100(60毫克)加入到乙醇(750微升)和水(750微升),加热到80℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与β-环糊精及糊精Maltrin M100的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例30
将依帕列净(30毫克)、微晶纤维素(3毫克)和羧甲基纤维素钠SCMS(3毫克)加入到水(30毫升),加热到100℃搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与琥珀酸及羧甲基纤维素钠SCMC的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例31
将依帕列净(5毫克)、微晶纤维素(5毫克)和聚环氧乙烷Polyox WSR301(30毫克)加入到甲醇(300微升)和水(60微升),60℃下搅拌混合均匀。将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与微晶纤维素及聚环氧乙烷Polyox WSR301的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例32
将依帕列净(30毫克)、微晶纤维素(20毫克、)聚乙二醇8000(20毫克)和聚乙烯醇EG-40(20毫克)加入到甲醇(300微升)和水(60微升),60℃下搅拌溶清,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与微晶纤维素、聚乙二醇8000及聚乙烯醇EG-40的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例33
将依帕列净(50毫克)、微晶纤维素(50毫克)和羟丙甲基纤维素醋酸酯琥珀酸酯Agoat MG(1克)加入到乙醇(10毫升)和水(2毫升),80℃下搅拌混合均匀,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与微晶纤维素及羟丙甲基纤维素醋酸酯琥珀酸酯Agoat MG的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例34
将依帕列净(50毫克)、藻酸盐(100毫克)和羧甲基乙基纤维素(1克)加入到乙醇(10毫升)和水(1毫升),80℃下搅拌混合均匀,将上述溶液在旋转蒸发器中缓慢浓缩除去溶剂,得到白色粉末状固体,即无定型依帕列净与藻酸盐及羧甲基乙基纤维素的固体分散体。该固体分散体的X-射线粉末衍射图中,扣除药用辅料的背景峰后无依帕列净晶型的特征峰。
实施例35:无定型依帕列净与羟丙基纤维素SSL及聚维酮K30的固体分散体的影响因素试验
材料:实施例1所得无定型依帕列净与羟丙基纤维素SSL及聚维酮K30的固体分散体
表1:
表1说明:无定型依帕列净与羟丙基纤维素SSL及聚维酮K30固体分散体在高温、高湿条件下,放置10天,有关物质无显著改变,无依帕列净结晶析出。
实施例36:无定型依帕列净与羟丙基纤维素SSL及聚维酮K30固体分散体的加速试验
材料:实施例1所得无定型依帕列净与羟丙基纤维素SSL及聚维酮K30的固体分散体
实验条件:温度40℃±2℃,湿度75%±5%
表2:
表2说明:无定型依帕列净与羟丙基纤维素SSL及聚维酮K30固体分散体在加速试验条件下,放置6个月,有关物质无显著改变,无依帕列净结晶析出。
本发明的依帕列净与药用辅料的无定型固体分散体,其溶出度明显增加,更有利于提高药物的生物利用度,使药物能够更好地发挥临床疾病治疗作用,该无定型物在加速试验条件下(40±2℃,湿度75%±5%),能保持良好的物理稳定性和化学稳定性。
- 还没有人留言评论。精彩留言会获得点赞!