使癌干细胞可视化并消除癌干细胞的组合物和方法与流程
相关申请的交叉引用
本申请要求于2009年2月26日递交的美国临时申请号61/155,624的权益,将其内容通过引用并入本文。
技术领域
本发明一般地涉及癌症诊断与治疗的领域,更具体而言涉及可用于消除HER2+乳腺癌细胞、不表达雌激素受体、孕酮受体或HER2/neu的三阴性乳腺癌细胞、和具有干细胞样特征的癌细胞的组合物和方法。本发明的组合物和方法还可用于处置转移乳腺癌和使患者体内的乳腺癌细胞可视化。
背景技术:
根据乳腺癌是否表达雌激素受体、孕酮受体和Her2/neu,可将其分为不同亚型。在开始癌症治疗之前,确定患者的癌症亚型很重要,这是因为有些药物靶向表达雌激素受体的癌细胞,而另一些药物靶向表达其他受体的癌细胞。已表明HER2/neu基因涉及哺乳动物肿瘤发生、肿瘤生长和转移。HER2/neu在20-30%的人乳腺癌中增多,并且HER2阳性亚型的乳腺癌与侵袭性转移疾病相关(Korkaya等人2008.Oncogene)。本发明通过提供可用于消除HER2阳性乳腺癌细胞的新型组合物和方法,可有助于治疗HER2阳性乳腺癌。
最近鉴定了一种新的乳腺癌细胞亚型。这些癌细胞由于不表达雌激素受体、孕酮受体或Her2/neu而被称为三阴性乳腺癌细胞(Dent等人2007.Clinical Cancer Research 13:4429-4434)。如Cancer Research UK(2007)所述,三阴性乳腺癌病例在所有乳腺癌病例中占约15%。与其他已知的乳腺癌亚型相比,三阴性亚型更具侵袭性、对标准治疗响应更低并且与整体患者预后更差有关(Dent等人2007.Clinical Cancer Research 13:4429-4434)。因此,需要诊断和治疗三阴性乳腺癌患者的组合物和方法。
近期研究表明,包括乳腺癌在内的多种癌症都是由小亚群的癌干细胞(CSC)因自我更新途径的失调而产生。已经报道具有干细胞样特征的乳腺癌细胞亚群为CD44和CD133阳性、CD24阴性并且表达醛脱氢酶1(ALDH1)(Crocker等人2008.J Cell Mol Med)。需要鉴定和消除具有干细胞样特征的乳腺癌细胞亚群的组合物和方法。
催乳素(“PRL”)是23-kDa的神经内分泌激素,在结构上与生长激素相关,并且以较低的程度与白介素家族的成员相关(Reynolds等人,1997,Endocrinol.138:5555-5560,Cunningham等人,1990,Science 247:1461-1465;Wells等人,1993,Recent Prog.Horm.Res.48:253-275)。催乳素受体存在于乳腺、卵巢、垂体腺、心脏、肺、胸腺、脾脏、肝脏、胰腺、肾脏、肾上腺、子宫、骨骼肌、皮肤和中枢神经系统的区域中。Mancini T.(2008),“Hyperprolactinemia and Prolactinomas,”Endocrinology&Metabolism Clinics of North America 37:67。当催乳素与其受体结合时,它引起它与另一催乳素受体二聚化,从而导致Janus激酶2的活化,Janus激酶2是启动JAK-STAT路径的酪氨酸激酶。催乳素受体的活化还导致有丝分裂原-活化蛋白激酶和Src激酶有丝分裂原的活化。Mancini,T.(2008).“Hyperprolactinemia and Prolactinomas,”Endocrinology&Metabolism Clinics of North America 37:67。“催乳素受体拮抗剂”是指干扰催乳素信号转导途径的催乳素形式。发明人为W.Chen和T.Wagner的美国专利申请2007/0060520之前已描述了这种催乳素受体拮抗剂。
技术实现要素:
本发明提供了一种用于抑制患者的不表达雌激素受体、孕酮受体或Her2/neu的乳腺癌三阴性细胞的生长的方法,所述方法包括对患者施用人催乳素受体拮抗剂。可用于该方法的人催乳素受体拮抗剂包括其中129位的甘氨酸被另一个氨基酸如精氨酸取代的那些人催乳素受体拮抗剂。可用于该方法的其他人催乳素受体拮抗剂包括其中129位的甘氨酸被以下氨基酸中的任一个取代的那些人催乳素受体拮抗剂:缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸、脯氨酸、酪氨酸、半胱氨酸、甲硫氨酸、精氨酸、组氨酸、色氨酸、苯丙氨酸、赖氨酸、天冬酰胺、谷氨酰胺、天冬氨酸或谷氨酸。
在另一些实施方式中,本发明提供了用于抑制乳腺癌患者的转移瘤的发展的方法,所述方法包括对患者施用人催乳素受体拮抗剂。可用于该方法的人催乳素受体拮抗剂包括其中129位的甘氨酸被另一个氨基酸如精氨酸取代的那些人催乳素受体拮抗剂。可用于该方法的其他人催乳素受体拮抗剂包括其中129位的甘氨酸被以下氨基酸中的任一个取代的那些人催乳素受体拮抗剂:缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸、脯氨酸、酪氨酸、半胱氨酸、甲硫氨酸、精氨酸、组氨酸、色氨酸、苯丙氨酸、赖氨酸、天冬酰胺、谷氨酰胺、天冬氨酸或谷氨酸。
本发明还涉及用于降低乳腺癌患者的醛脱氢酶1(ALDH1)的活性的方法,所述方法包括对患者施用人催乳素受体拮抗剂。可用于该方法的人催乳素受体拮抗剂包括其中129位的甘氨酸被另一个氨基酸如精氨酸取代的那些人催乳素受体拮抗剂。可用于该方法的其他人催乳素受体拮抗剂包括其中129位的甘氨酸被以下氨基酸中的任一个取代的那些人催乳素受体拮抗剂:缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸、脯氨酸、酪氨酸、半胱氨酸、甲硫氨酸、精氨酸、组氨酸、色氨酸、苯丙氨酸、赖氨酸、天冬酰胺、谷氨酰胺、天冬氨酸或谷氨酸。
本发明还涉及用于减少乳腺癌患者的癌干细胞的数量的方法,所述方法包括对患者施用人催乳素受体拮抗剂。可用于该方法的人催乳素受体拮抗剂包括其中129位的甘氨酸被另一个氨基酸如精氨酸取代的那些人催乳素受体拮抗剂。可用于该方法的其他人催乳素受体拮抗剂包括其中129位的甘氨酸被以下氨基酸中的任一个取代的那些人催乳素受体拮抗剂:缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸、脯氨酸、酪氨酸、半胱氨酸、甲硫氨酸、精氨酸、组氨酸、色氨酸、苯丙氨酸、赖氨酸、天冬酰胺、谷氨酰胺、天冬氨酸或谷氨酸。本发明的方法中的任一个均可通过与选自毒素、放射性同位素、荧光染料和蛋白的试剂缀合的催乳素受体拮抗剂来实施。在另一些实施方式中,可以通过将催乳素受体拮抗剂与化学治疗剂同时施用或依次施用来进行本发明的方法。这种化学治疗剂可以包括以下化合物中的任一个或它们的组合:全反式视黄酸;阿扎胞苷;硫唑嘌呤;博来霉素;卡铂;卡培他滨;顺铂;苯丁酸氮芥;环磷酰胺;阿糖胞苷;道诺霉素;多西他赛;去氧氟尿苷;阿霉素;表柔比星;埃博霉素;依托泊苷;氟尿嘧啶;吉西他滨;羟基脲;伊达比星;伊马替尼;氮芥;巯基嘌呤;甲氨蝶呤;米托蒽醌;奥沙利铂;紫杉醇;培美曲塞;替尼泊苷;硫鸟嘌呤;戊柔比星;长春碱;长春新碱;长春地辛和长春瑞滨。
可通过各种施用途径将催乳素受体拮抗剂递送至患者,所述各种施用途径包括但不限于胃肠外、皮下、腹膜内、静脉内、淋巴管内、鞘内、心室内或肺内。在本发明的方法中,可以在乳腺癌切除之前和/或之后施用催乳素受体拮抗剂。
本发明还涉及定位患者体内的癌细胞并且鉴定转移的方法。为了实现该目的,将与放射性同位素、荧光染料或其他标记试剂缀合的催乳素受体拮抗剂施用于患者,然后对该患者进行鉴定该拮抗剂定位区域的程序。可用于定位缀合催乳素受体的程序包括计算机断层扫描程序、计算机断层扫描程序、磁共振成像和核磁共振成像。
附图说明
图1.用催乳素受体拮抗剂G129R治疗三阴性乳腺癌细胞使ALDH1活性降低。
图2.用催乳素受体拮抗剂G129R治疗负荷HER2+/neu乳腺肿瘤的小鼠使肿瘤细胞的ALDH1活性降低。
图3.用催乳素受体拮抗剂G129R治疗原发HER2+/neu乳腺癌细胞使ALDH1活性降低。
图4.催乳素拮抗剂G129R抑制携带HER2+/neu乳腺肿瘤的哺乳动物中转移瘤的发展。
图5.催乳素拮抗剂G129R抑制哺乳动物中HER2+/neu肿瘤的生长。
图6.催乳素拮抗剂G129R的治疗抑制哺乳动物原发和继发肿瘤中HER2+/neu蛋白的磷酸化。
图7.体内HER2+肿瘤对催乳素拮抗剂G129R治疗的剂量依赖性响应。
图8.体内HER+肿瘤对催乳素拮抗剂G129R治疗的时间依赖性响应。
图9.人催乳素的氨基酸序列。
具体实施方式
人催乳素的氨基酸序列如图9所示。术语“催乳素(PRL)”在本文中指人和非人动物形式的激素催乳素(还可见Cooke等人,J.Biol.Chem.,256:4007(1981);Cooke等人,J.Biol.Chem.,225:6502(1980);Kohmoto等人,Eur.J.Biochem.,138:227(1984);Tsubokawa等人,Int.J.Peptide Protein Res.,25:442(1985);Bondar等人,GenBank登录号#X63235(1991);Sasavage等人,J.Biol.Chem.257:678(1982);Miller等人,Endocrinol.107:851(1980);Li等人,Arch.Biochem.Biophys.141:705(1970);Li,Int.J.Peptide Protein Res.,8:205(1976);Martinant等人,Biochim.Biophys.Acta,1077:339(1991);Lehrman等人,Int.J.Peptide Protein Res.,31:544(1988);Li等人,Int.J.Peptide Protein Res.,33:67(1989);Hanks等人,J.Mol.Endocrinol.,2:21(1989);Watahiki等人,J.Biol.Chem.,264:5535(1989);Karatzas等人,Nucl.Acids Res.,18:3071(1990);Yasuda等人,Gen.Comp.Endocrinol.,80:363(1990);Noso等人,Int.J.Peptide Protein Res.,39:250;Buckbinder等人,Proc.Natl.Acad.Sci.U.S.A.,90:3820(1993);Takahashi等人,J Mol.Endocrinol.,5:281;Yamaguchi等人,J.Biol.Chem.,263:9113(1988);Rentler-Delrue等人,DNA,8:261;Yasuda等人,Gen.Comp.Endocrinol.,66:280(1987);Chang等人,GenBank登录号#X61049(1991);Chang等人,GenBank登录号#X61052(1991);Yasuda等人,Arch.Biochem.Biophys.,244:528(1986);Kuwana等人,Agric.Biol.Chem.,52:1033(1988);Song等人,Eur.J.Biochem.,172:279(1988);Mercier等人,DNA 8:119(1989))。
术语“催乳素受体拮抗剂”是指干扰催乳素信号转导途径的催乳素形式。优选的催乳素受体拮抗剂包括其中至少一个氨基酸通过插入、缺失和/或取代而由其天然存在序列改变的催乳素。术语G129R指其中129位甘氨酸由精氨酸取代的例如图9所示的催乳素受体拮抗剂。
将催乳素受体拮抗剂拮抗PRL对其受体的作用的能力定义为该变体抑制由PRL介导的效应的能力。催乳素受体拮抗剂包括公开的美国专利申请号2005/0271626中描述的那些催乳素受体拮抗剂,将其内容以引用方式整体并入本文。
在PRL和PRL变体都存在时,通过测定催乳素变体(“PRL变体”)对PRL经由其受体发挥作用的能力进行阻断的能力,可以鉴定催乳素受体拮抗剂。PRL经由其受体发挥作用的能力可通过监测细胞增殖的变化以及MAP-激酶和HER2/neu信号转导途径中下游靶标的磷酸化/活化来测定。
可以将其中129位的甘氨酸残基被另一个氨基酸替换的催乳素变体用于本发明的方法中。以缩写形式G 129*(其中*是除甘氨酸之外的天然存在或合成氨基酸)表示的该取代可以是天然存在序列的唯一变化,也可以是多个改变(包括其他氨基酸的插入、缺失和/或取代)中的一个。取代氨基酸可以是中性-极性氨基酸,例如丙氨酸、缬氨酸、亮氨酸、异亮氨酸、苯丙氨酸、脯氨酸、甲硫氨酸;中性非极性氨基酸,例如丝氨酸、苏氨酸、酪氨酸、半胱氨酸、色氨酸、天冬酰胺、谷氨酰胺、天冬氨酸;酸性氨基酸,例如天冬氨酸或谷氨酸;以及碱性氨基酸,例如精氨酸、组氨酸或赖氨酸。在本发明优选实施方式中,hPRL的129位甘氨酸可以被缬氨酸、亮氨酸、异亮氨酸、丝氨酸、苏氨酸、脯氨酸、酪氨酸、半胱氨酸、甲硫氨酸、精氨酸、组氨酸、色氨酸、苯丙氨酸、赖氨酸、天冬酰胺、谷氨酰胺、天冬氨酸或谷氨酸取代。在本发明的一个实施方式中,该取代是以精氨酸替换129位甘氨酸(G129R)。在另一实施方式中,本发明提供了其中129位甘氨酸缺失的催乳素变体。
可以将催乳素受体拮抗剂与作为融合蛋白一部分的另一蛋白连接。例如,可将催乳素拮抗剂与白介素2、绿色荧光蛋白、β-半乳糖苷酶或选自例如美国专利7,425,535所述的那些蛋白的孔形成蛋白连接。在另一实施方式中,催乳素受体拮抗剂与化合物缀合。这种化合物包括、但不限于荧光染料、放射性同位素或细胞毒素,所述细胞毒素例如能够引发细胞凋亡的小分子。
本发明的催乳素受体拮抗剂可通过化学合成或通过重组DNA技术制备。通常,可通过以下方式制备PRL的cDNA:利用标准PCR扩增技术,以从产生PRL的细胞(例如垂体细胞)制备的RNA或cDNA为模板,根据已知PRL核酸或氨基酸序列设计寡核苷酸引物。制备编码HPRL的cDNA的非限制性实例如公开的美国专利7,115,556(Wagner等人)所述。然后可随机或通过定点突变在PRL cDNA中引入改变。
在通过重组技术制备催乳素受体拮抗剂的时候,可以将编码PRL变体的核酸引入表达载体,并可操纵地连接于合适的启动子/增强子序列。表达载体还可以含有一个或多个辅助PRL变体表达的元件,包括转录终止位点、多聚腺苷酸位点、核糖体结合位点、信号序列等。合适的表达系统包括哺乳动物细胞、昆虫细胞、植物细胞、酵母细胞、粘液菌、和包括转基因植物和转基因动物在内的生物体。适合的表达载体包括单纯疱疹病毒类载体如pHSV1(Geller等人,Proc.Natl.Acad.Sci.U.S.A.87:8950-8954(1990));逆转录病毒载体如MFG(Jaffee等人,Cancer Res.53:2221-2226(1993)),特别是莫洛尼逆转录病毒载体如LN、LNSX、LNCX、LXSN(Miller和Rosman,Biotechniques,7:980-989(1989));牛痘病毒载体如MVA(Sutter和Moss,Proc.Natl.Acad.Sci.U.S.A.89:10847-10851(1992));腺病毒载体如pJM17(Ali等人,Gene Therapy 1:367-384(1994);Berker,Biotechniques 6:616-624(1988);Wand和Finer,Nature Medicine 2:714-716(1996));腺相关病毒载体如AAV/neo(Mura-Cacho等人,J.Immunother.,11:231-237(1992));慢病毒载体(Zufferey等人,Nature Biotechnology 15:87l-875(1997));质粒载体如pCDNA3和pCDNAl(InVitrogen)、pET 11a、pET3a、pET11d、pET3d、pET22d、和pET12a(Novagen);质粒AH5(其含有SV40起点和腺病毒主要后期启动子)、pRC/CMV(InVitrogen)、pCMU II(Paabo等人,EMBO J.,5:1921-1927(1986)),pZipNeo SV(Cepko等人,Cell,37:1053-1062(1984))。pSR.α.(DNAX,Palo Alto,Calif.)和pBK-CMV;以及杆状病毒表达载体(O′Reilly等人,BACULOVIRUS EXPRESSION VECTORS,Oxford University Press(1995)),例如p2Bac(InVitrogen)。
随后可通过标准技术对重组表达系统产生的催乳素受体拮抗剂进行纯化,所述标准技术包括电泳、色谱(包括亲和色谱)、和超滤。可以制备与放射性同位素或荧光染料缀合的肽形式的催乳素受体拮抗剂。可以在体外转录/翻译系统中合成生物素化或放射性标记的催乳素受体拮抗剂,在所述体外转录/翻译系统中将变体基因克隆到载体中的SP6启动子下,随后通过利用SP6转录酶和兔网织红细胞在生物素或放射性标记物的存在下制备蛋白。体外转录/翻译系统可商购自QIAGEN。作为另一种选择,可以在细菌细胞或哺乳动物细胞中产生缀合的催乳素受体拮抗剂,所述细菌细胞或哺乳动物细胞在放射性标记的、荧光标记的或生物素化的氨基酸存在下培养。也可利用相关领域技术人员公知的其他合成标记蛋白的方法来制备与放射性同位素、荧光染料、顺磁标签或生物素缀合的催乳素受体拮抗剂。
本发明提供了其中可利用催乳素受体拮抗剂抑制三阴性乳腺癌细胞或Her2+乳腺癌细胞增殖的方法和组合物。在一些实施方式中,通过获取生物活检样并以标记物对所获乳腺癌细胞染色,所述标记物可对癌细胞是否表达雌激素受体、HER2+和/或孕酮受体进行可视化,从而对乳腺癌患者进行诊断,以确定其乳腺癌是否为三阴性乳腺癌亚型或HER2阳性亚型。如果确定患者携带三阴性乳腺癌细胞或HER2+乳腺癌细胞,则用催乳素受体拮抗剂治疗患者。
为了确定用于治疗的催乳素受体拮抗剂的有效量,可使用标准方法,如剂量-响应测试。然后以催乳素受体拮抗剂对患者按日进行治疗。可以监测患者对治疗的响应,三阴性乳腺癌细胞或HER2阳性细胞数量减少表明对治疗有正面响应。每轮治疗之后根据需要和总体改善,还可以对患者进行后续轮次的治疗。
本发明还提供了其中可使用催乳素受体拮抗剂治疗卵巢癌、子宫癌(子宫内膜癌)、和宫颈癌的方法和组合物。为了确定用于治疗的催乳素受体拮抗剂的有效量,可使用标准方法,如剂量-响应测试。每轮治疗之后根据需要和总体改善,还可以对患者进行后续轮次的治疗。
本发明还提供了其中可使用催乳素受体拮抗剂消除具有转移瘤的患者的继发肿瘤细胞的方法和组合物。在所述方法中,在乳腺癌外科手术过程中从患者肿瘤取活检物。随后分析活检物,从而确定患者癌症亚型是否是三阴性或HER2阳性。随后使患者从其外科手术中恢复,并对其进行催乳素受体拮抗剂治疗,例如为期2周的G129R治疗。1至2周后,对患者再进行数轮治疗。催乳素受体拮抗剂治疗可以与其他治疗方法如化学疗法和/或放射同时施用。
本发明还提供了其中可使用催乳素受体拮抗剂消除具有干细胞样特征的乳腺癌细胞的方法和组合物。在一些实施方式中,如上所述对患者进行催乳素受体拮抗剂治疗。
本发明还提供了其中可使用催乳素受体拮抗剂对患者的具有干细胞样特征的乳腺癌细胞进行可视化的方法和组合物。在一些实施方式中,将催乳素拮抗剂首先与可检测标记物如荧光染料或放射性同位素缀合,然后施用于患者。然后通过本领域已知程序中的任一种将拮抗剂在患者体内的定位可视化。所述程序例如包括计算机断层扫描程序;磁共振成像和核磁共振成像。
在本发明的方法中,可以将催乳素受体拮抗剂与其他适合治疗乳腺癌的试剂相继施用或在组合治疗方案中一起施用。作为非限制性实例,用于组合方案的其他试剂可包括化学治疗剂、使HER2/neu信号转导途径失活的试剂如贺赛汀、抗雄激素和/或抗雌激素,如他莫昔芬。
对于治疗应用,本发明的组合物可以以药学可接受剂型施用于哺乳动物、优选人,所述药学可接受剂型包括可以作为静脉内推注施用或经一段时间连续输注、或通过肌肉内、腹膜内、脑脊腔内、皮下、动脉内、滑膜内、鞘内、口服、局部或吸入途径施用的那些剂型。本发明的组合物还适合于经肿瘤内、肿瘤周围、病损内或病损周围途径施用,以便发挥局部的和全身的效果。预计腹膜内施用对各种癌症和转移病灶的治疗特别有用。
可根据已知的制备药用组合物的方法来配制本发明的组合物,通过所述方法将组合物试剂组合于与药学可接受载体载质(carrier vehicle)的混合物中。包括其他人类蛋白如人血清白蛋白在内的适合的载质及其配制,在例如REMINGTON′S PHARMACEUTICAL SCIENCES(16版,Osol,A编,Mack,Easton Pa.(1980))中有所描述。为了形成适合有效施用的药学可接受组合物,所述组合物含有有效量的本发明的蛋白中的一种或多种,以及适合量的载体载质。更具体而言,有效剂量(量)是指抑制HER2+乳腺癌细胞或三阴性乳腺癌细胞增殖所需的催乳素受体拮抗剂的量。可以根据实施例8所述的剂量-响应和时间-响应测试确定有效量。治疗有效量的确定特别取决于药物的毒性和功效等因素。可以利用本领域公知的方法和在先文献中的方法来确定毒性。可利用相同的指导结合以下实施例所述方法来确定功效。因此,药学有效量就是指被临床医师视为毒性可耐受并仍然有功效的量。功效例如可通过靶向的肿瘤块的质量减少而测定。
可以用催乳素受体拮抗剂组合物治疗乳腺癌、卵巢癌、子宫癌(子宫内膜癌)或宫颈癌的患者。随后,可以通过监测患者癌细胞中ALDH1活性的下降来监测患者的具有干细胞样特征的癌细胞的数量的减少。之后可重复进行催乳素受体拮抗剂治疗的轮次,从而进一步消除具有干细胞样特征的癌细胞。在本发明的一个实施方式中,可对患者处以1至6轮治疗,每轮由1至10天组成。
可使用催乳素受体拮抗剂对具有干细胞样特征的乳腺癌、卵巢癌、子宫癌(子宫内膜癌)或宫颈癌细胞进行可视化。在一个实施方式中,可以施用与绿色荧光蛋白融合的催乳素受体拮抗剂。在其他实施方式中,可以将催乳素受体拮抗剂与荧光染料或放射性同位素缀合,并施用于患者。随后对催乳素受体拮抗剂在患者体内的定位进行监测。
可以利用一种或多种生理可接受的载体或赋形剂通过常规方式来配制根据本发明所用的组合物。因此,可将其配制为用于通过吸入或吹入施用(通过嘴或鼻),或口服、口颊、胃肠外或直肠施用。
可以对口服施用制剂进行适当配制,从而提供活性化合物的控释。对于口颊施用,组合物可以为常规方法配制的片剂或锭剂的形式。对于吸入施用,可以将根据本发明的组合物以气雾喷剂方式由加压包或喷雾器按常规方式递送,并使用适当的推进剂,例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其他适当气体。在加压气雾剂情况中,可以通过设置可递送计量量的阀来确定剂量单位。可以配制含有化合物和适当粉末基料如乳糖或淀粉的粉末混合物的胶囊和盒(例如明胶胶囊和盒),以用于吸入器或吹入器。
可以将本发明的组合物配制用于通过注射进行胃肠外施用,例如可通过推注或连续输注。注射用制剂可以以单位剂量形式存在,例如在安培瓶或在多剂量容器中,并添加有防腐剂。组合物可以为在油性或水性载质中的悬浮液、溶液或乳液等形式,并且可含有配制剂如悬浮剂、稳定剂和/或分散剂。作为另一种选择,活性成分可以为粉末形式,以便在使用前用适当载质如无菌无热源水配制。还可以将组合物配制为直肠用组合物,例如如含有常规栓剂基质(如可可油或其他甘油酯)的栓剂或灌肠保留剂。除了上述制剂以外,还可以将本发明的组合物配制为长效制剂(depot preparation)。这种长时间作用制剂可通过植入(例如皮下或肌肉内植入)或肌肉内注射来施用。因此,例如可以将使HER2/neu信号转导途径失活的催乳素变体和/或试剂与适当聚合物或疏水材料(如作为在可接受油中的乳液)或离子交换树脂配制,或配制为微溶衍生物,例如微溶盐。
如需要,该组合物可存在于可含有一个或多个包含活性成分的单位剂量形式的包装或分配装置中。所述包装例如可以具有金属箔或塑料箔,如泡罩包装。所述包装或分配装置可带有施用说明书。
此外,还可以对本发明的组合物进行修饰,从而具有更长清除率,因而通过保护组合物免受受试者的免疫反应和其他清除基质的破坏而增加生物利用度。例如,PEG化的化合物显示降低的免疫原性和抗原性,并在血流内的循环比未缀合的蛋白显著延长。可通过本领域已知方法将PEG(聚乙二醇)聚合物链与催乳素变体/催乳素受体拮抗剂连接,例如通过Roberts等人,Adv.Drug Del.Rev.,54(4):459-76(2002)所述的PEG化程序。但其他可延长本发明治疗组合物的清除半衰期的试剂是本领域技术人员已知的并且也包含在本发明中。
实际上,还可以用羟乙基淀粉(HES)修饰本文所述的组合物。HES是天然存在的支链淀粉的衍生物,并在体内被α淀粉酶降解。本领域已知制备HES-蛋白缀合物的方法。例如见EP 1398322、DE 2616086和DE 2646854。
为了延长本发明组合物的清除时间并增加其半衰期,本发明还包括将催乳素受体拮抗剂与血清白蛋白连接的试剂。虽然美国专利公开2007/0160534(将其通过引用并入本文)披露了这种试剂,还认为其他对可缀合本发明的催乳素受体拮抗剂的血清白蛋白有亲和性的肽配体适合用于此处。
参考以下实施例对本发明进行进一步描述,这些实施例仅用于说明目的。本发明不限于这些实施例,而是包含了因本文提供的教导而显而易见的各种变化形式。
实施例1
人催乳素拮抗剂G129R的制备
利用逆转录(RT)和之后的聚合酶链式反应(PCR)成功克隆了人PRL。简单来说,将人垂体polyA RNA(CloneTech,Inc.Palo Alto,Calif.)用作模板。由hPRL cDNA的距终止密码子(TAA)2个碱基处开始设计HPRL反义引物(5′-GCTTAGCAGTTGTTGTTGTG-3′;SEQ ID NO:1),并从ATG起设计正义引物(5′-ATGAACATCAAAGGAT-3′;SEQ ID NO:2)。利用Perkin-Elmer Cetus,Inc.(Norwalk,Conn.)的试剂盒进行RT/PCR。利用经修饰的T7 DNA聚合酶(Sequenase,United States Biochemical)通过双脱氧链终止法测定所得hPRL的核苷酸序列,发现其与GenBank中报导的相似,不同之处在于导致密码子21处的沉默突变的一个碱基差异(CTG->CTC)。包括pUCIG-Met表达载体制备在内的克隆过程的示意图在公开的美国申请号20030022833号(Wagner等人)中有所总结,将该文献通过引用整体并入本文。
将含有hPRL cDNA和M13 F1复制起点的亲本质粒转化到E.coli(CJ236)中。利用辅助噬菌体M13k07从所转化的CJ236菌中分离出含有尿苷的单链质粒DNA。通过在70℃加热5分钟,然后缓慢冷却,将6pmol含有指导G129R突变的寡核苷酸与0.2pmol单链DNA在退火缓冲液(200mM Tris-HCl,20mM MgCl2,100mM NaCl)中退火。用单链DNA为模板,以T4 DNA聚合酶催化,将编码G129R突变的寡核苷酸(5′-CGGCTCCTAGAGAGGATGGAGCT-3′;SEQ ID NO:3)用于引发DNA互补链的合成。合成后,使用双链DNA来转化E.coli(DH5a)。分离单克隆,并通过DNA核苷酸测序来筛选hPRL-G129R。
将hPRL和G129R编码核酸各自插入哺乳动物细胞表达载体中,所述哺乳动物细胞表达载体之中cDNA的转录受到小鼠金属硫蛋白增强子/启动子序列和bGH poly A添加信号的控制(Chen等人,J.Biol.Chem.,266:2252-2258(1991);Chen等人,Endocrinol.,129:1402-1408(1991);Chen等人,Mol.Endocrinol.,5:1845-1852(1991);Chen等人,J.Biol.Chem.,269:15892-15897(1994))。为了建立产生hPRL和hPRLA的稳定小鼠L细胞系,将小鼠L细胞[胸苷激酶-阴性(TK),且腺苷磷酸核糖基转移酶-阴性(APRT)]选作体外表达体系。制备了表达HPRL的稳定细胞系(其被用作阳性对照)和人催乳素拮抗剂(约5-10mg/l/24h/百万个细胞)。
利用Chen等人,J.Biol.Chem.269:15892-15897(1994)所述技术,使用膜超滤从条件细胞培养基中部分纯化和浓缩hPRL和人催乳素拮抗剂。分离基于相对分子尺寸和膜的孔尺寸。超滤膜获自Amicon,Inc.(Northorough,Mass.)。使用了两种膜,YM10和YM100。首先,使用具有Amicon YM100的200ml超滤装置,在20psia跨膜压下从培养基中除去大杂质。将透过物(hPRL回收为>90%)用于第二过滤规程,该第二过滤规程用YM10膜来减少溶液体积,从而浓缩蛋白。利用Diagnostic Products Corp.(Los Angeles,Calif.)的免疫放射计量测试(IRMA)试剂盒来确定HPRL或人催乳素拮抗剂的浓度。
实施例2
用催乳素受体拮抗剂G129R治疗三阴性乳腺癌细胞可降低细胞的ALDH1活性。
将人三阴性乳腺癌细胞MDA-MB-231和对照乳腺癌细胞T-47D以1x105个细胞/孔铺于6孔板中,并使其在生长培养基中贴壁24小时,然后进行2小时的无血清培养基消耗。随后在不含(对照)或含有催乳素(100ng/ml)或G129R(10ug/ml)的情况下培养细胞3天。然后收集细胞,并通过ALDEFLUORR荧光测试(StemCell Technologies)测定ALDH-1活性(该酶是癌干细胞的标记物。见Ginestier等人,Cell Stem Cell 20071(5):555-567)。如图1所示,催乳素拮抗剂G129R治疗显著降低了三阴性乳腺癌细胞中的ALDH1活性。
实施例3
催乳素受体拮抗剂治疗降低肿瘤细胞中的ALDHl活性
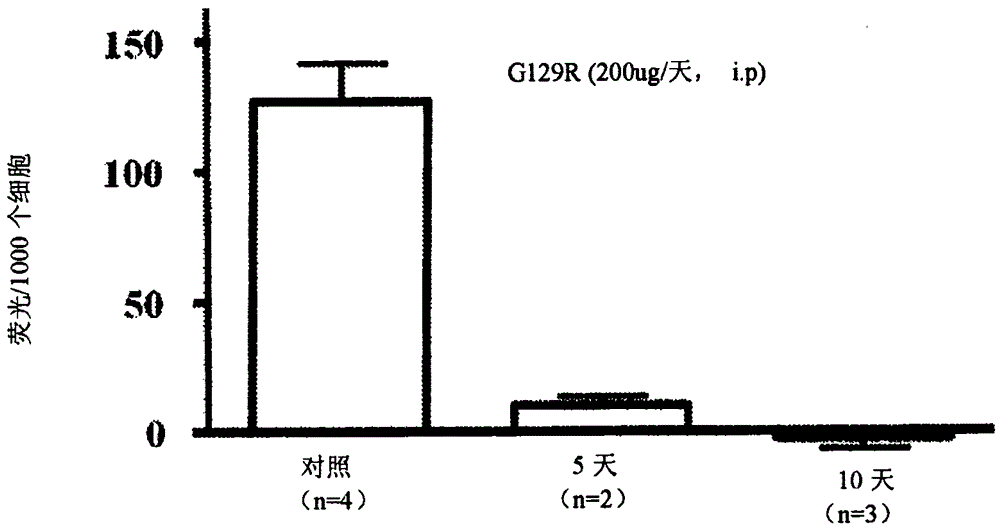
MMTV/neu转基因小鼠携带由小鼠乳腺肿瘤病毒(MMTV)启动子驱动的活化的c-neu癌基因,并发展为乳腺HER2+腺癌(Muller等人1988.Cell.54(1):105-15)。对携带乳腺肿瘤的MMTV/neu转基因小鼠以PBS(n=4)或G129R治疗5天(n=2)或10天(n=3)(10mg/kg,i.p.)。在治疗结束时分离肿瘤,消化为单细胞悬浮液,并比较ALDH1活性水平。从图2可见,在分离自G129R治疗动物的癌细胞中,ALDH1活性显著降低。实际上在以G129R治疗10天的癌细胞中未检测到ALDH1活性。
实施例4
催乳素受体拮抗剂对癌细胞的治疗使ALDH1活性降低
从MMTV/neu转基因小鼠中分离原发乳腺肿瘤细胞,并在G129R(10ug/ml)或PRL(100ng/ml)存在下培养24、48、72、或96小时。具体而言,将取自MMTV-neu小鼠的肿瘤消化为单细胞悬浮液,并以1x105个细胞/ml铺在12孔板中。使细胞在生长培养基中贴壁24小时,然后进行2小时的无血清培养基消耗。随后不用(对照)催乳素或用催乳素(100ng/ml)或G129R(10ug/ml)处理细胞。处理24、48、72、或96小时后收集细胞,并测定ALDH-1活性。由图3可见,对暴露于G129R的原发MMTV/neu肿瘤细胞进行的时间过程研究揭示出ALDH1活性的稳定下降,最终水平显著低于对照(96小时减少92.3%)。
实施例5
催乳素拮抗剂抑制哺乳动物转移瘤的发展
从MMTV/neu转基因小鼠中分离原发乳腺肿瘤。以PBS或G129R(200ug/天,i.p.)对小鼠治疗超过40天。当继发肿瘤(复发)生长至一定尺寸,处死小鼠,切取肺并在包氏固定剂中固定以便观察转移。图4显示了在G129R治疗组和对照组中具有肺转移瘤的动物的百分比(应用卡方检验确定显著性)。由图4可见,与未治疗的对照组相比,G129R治疗组中具有肺转移瘤的动物的百分比显著降低**(P<0.05)。
实施例6
催乳素拮抗剂抑制哺乳动物的肿瘤生长
在两个分开的实验中,从雌性转基因小鼠中分离肿瘤,将其切为等尺寸片并植入到年龄匹配的受体雌性转基因小鼠。植入后10天,将小鼠随机分为对照组(n=3-4)或G129R治疗组(n=4,10mg/kg,i.p.每日进行)。对小鼠以每日为基础进行50天治疗,并监测对照组和治疗组的肿瘤体积。如图5所示,与对照组相比,两个G129R治疗组中最终肿瘤体积小67.1%(±9.18SEM)或71.5%(±14.9SEM)。从图5中还可以看出,与对照组相比,G129R治疗组中最终肿瘤重量减少61.3%(±18.4SEM)或62.5%(±7.07SEM)。
实施例7
催乳素拮抗剂治疗抑制哺乳动物原发和继发肿瘤中HER2+/neu蛋白的磷酸化
每天以PBS(对照)或200ug的G129R对MMTV/neu小鼠(携带原发和继发肿瘤)治疗5天。第5次治疗后24小时取出原发和继发肿瘤,并进行处理以用于蛋白质印记。如图6所示,在分离自G129R治疗动物的原发肿瘤中,磷酸化Neu蛋白显著减少。G129R治疗动物的继发肿瘤也显示出neu磷酸化的降低。然后,在G129R治疗小鼠的继发肿瘤中仍可检测出一些磷酸化neu蛋白(见图6),提示催乳素拮抗剂G129R治疗可能以不同方式影响原发和继发肿瘤。
实施例8
体内HER+肿瘤对催乳素拮抗剂G129R治疗的剂量-依赖响应和体内HER+肿瘤对催乳素拮抗剂G129R治疗的时间-依赖响应
为确定降低HER+/neu体内磷酸化所需的G129R的最小剂量,由MMTV/neu转基因小鼠的肿瘤中移取活检物,将其用作自我对照基线。然后以以下浓度的PBS或G129R对小鼠经腹膜内治疗10天:每日50、100、200或400ug(2.5mg/kg/天、5mg/kg/天;10mg/kg/天;或20mg/kg/天的G129R)。然后收集肿瘤,并对其处理以进行蛋白质印记。然后以检测磷酸化HER2/neu蛋白的抗体和抗β-微管蛋白的抗体对蛋白质印记进行探测,β-微管蛋白是凝胶上样及蛋白质印记转移的对照。由图7可见,在G129R剂量为5mg/kg/天那样低时,就观察到G129R对HER+/neu磷酸化的抑制效果。
为确定G129R治疗的最佳长度,用200ug G129R对携MMTV瘤小鼠治疗5天或10天。最后一次治疗后24小时取出肿瘤,并对其处理以用于蛋白质印记分析。由图8可见,在接受5天G129R(10mg/kg/天,i.p.)治疗的14只小鼠中,5只小鼠的肿瘤与对照小鼠(n=8)相比显示出磷酸化HER2/neu蛋白的显著降低,4只为中度响应,可观察到磷酸化HER2/neu蛋白的降低,而5只没有响应。由图8可见,在接受10天G129R(10mg/kg/天,i.p.)治疗的10只小鼠中,3只小鼠的肿瘤具有高度响应,在这些肿瘤中实际上检测不到磷酸化HER2/neu,5只小鼠为中度响应,而2只没有响应。
序列表
<110> Oncolix, Inc.
<120> 使癌干细胞可视化并消除癌干细胞的组合物和方法
<130> 03153.0015.00PC00
<150> 61/155,624
<151> 2009-02-26
<160> 4
<170> PatentIn version 3.4
<210> 1
<211> 20
<212> DNA
<213> 人工
<220>
<223> 合成寡核苷酸
<400> 1
gcttagcagt tgttgttgtg 20
<210> 2
<211> 16
<212> DNA
<213> 人工
<220>
<223> 合成寡核苷酸
<400> 2
atgaacatca aaggat 16
<210> 3
<211> 23
<212> DNA
<213> 人工
<220>
<223> 合成寡核苷酸
<400> 3
cggctcctag agaggatgga gct 23
<210> 4
<211> 199
<212> PRT
<213> 人
<400> 4
Leu Pro Ile Cys Pro Gly Gly Ala Ala Arg Cys Gln Val Thr Leu Arg
1 5 10 15
Asp Leu Phe Asp Arg Ala Val Val Leu Ser His Tyr Ile His Asn Leu
20 25 30
Ser Ser Glu Met Phe Ser Glu Phe Asp Lys Arg Tyr Thr His Gly Arg
35 40 45
Gly Phe Ile Thr Lys Ala Ile Asn Ser Cys His Thr Ser Ser Leu Ala
50 55 60
Thr Pro Glu Asp Lys Glu Gln Ala Gln Gln Met Asn Gln Lys Asp Phe
65 70 75 80
Leu Ser Leu Ile Val Ser Ile Leu Arg Ser Trp Asn Glu Pro Leu Tyr
85 90 95
His Leu Val Thr Glu Val Arg Gly Met Gln Glu Ala Pro Glu Ala Leu
100 105 110
Leu Ser Lys Ala Val Glu Ile Glu Glu Gln Thr Lys Arg Leu Leu Arg
115 120 125
Gly Met Glu Leu Ile Val Ser Gln Val His Pro Glu Thr Lys Glu Asn
130 135 140
Glu Ile Tyr Pro Val Trp Ser Gly Leu Pro Ser Leu Gln Met Ala Asp
145 150 155 160
Glu Glu Ser Arg Leu Ser Ala Tyr Tyr Asn Leu Leu His Cys Leu Arg
165 170 175
Arg Asp Ser His Lys Ile Asp Asn Tyr Leu Lys Leu Leu Lys Cys Arg
180 185 190
Ile Ile His Asn Asn Asn Cys
195
- 还没有人留言评论。精彩留言会获得点赞!