一种用于肿瘤成像与治疗的纳米材料及其制备方法与流程
本发明涉及一种纳米材料,尤其涉及一种用于肿瘤成像与治疗的纳米材料。
背景技术:
《世界癌症报告》指出,全球癌症的发病率与死亡率呈持续上升的趋势。中国2015年约有430万人确诊癌症,280万人死于癌症,平均每天7500人。癌症给患者、家庭、社会带来极大的痛楚,因此,癌症的诊断和治疗已成为目前医学研究中的当务之急。
在癌症的成像诊断方面,磁共振成像(Magnetic Resonance Imaging,MRI)是一种非损伤性的成像方式,组织分辨率高,能实现全方位的成像。此外,上转换发光材料采用近红外光为激发源,光的穿透深度较深,没有生物背景荧光,生物相容性好,因此上转换发光(upconversion luminescence,UCL)成像也具有较好的生物应用前景。光热治疗(photothermal therapy,PTT)是癌症治疗一种全新的方法,不同于化疗和光动力疗法,它具有毒副作用较小,能治疗不同类型的肿瘤,治疗具有可重复性等优点。
针对目前临床上癌症诊断和治疗相互独立,治疗周期长,费用高,化学治疗、放射治疗、手术治疗等治疗手段副作用大等问题,为提高治疗的精准度,缩短治疗周期,减少对正常组织的损伤,将多模式成像与治疗相结合,制备具有诊疗一体化功能的新型纳米材料,成为癌症治疗的新方向。由于不同类型材料的物理化学性质迥异,合成多功能复合材料的难度增大。因此,研究制备多功能纳米诊疗剂,实现肿瘤诊断治疗一体化的纳米材料具有重要的理论意义和临床应用价值。
技术实现要素:
针对上述不足,本发明的目的在于提供一种用于肿瘤成像与治疗的纳米材料及其制备方法,克服目前癌症诊疗过程中所遇到的诊断治疗互相独立,肿瘤治疗药物靶向效果差,副作用大等问题,实现T1/T2成像与上转换荧光成像的双模成像与通过热效应进行肿瘤治疗,实现诊疗一体化。
本发明为达到上述目的所采用的技术方案是:
一种用于肿瘤成像与治疗的纳米材料的制备方法,包括以下步骤:
(1)以油酸钠与氯化铁为原料,溶于于乙醇、去离子水与环己烷的混合液,在60-80℃冷凝回流的环境中反应3-6小时,加水振荡反复洗涤后,弃去油相,于100-140℃下真空干燥一夜,得油酸铁;
(2)采用热裂解法,以步骤(1)所得油酸铁为原料,溶于油酸与十八烯的混合液中、在真空100-120℃的环境下加热20-40min,其后充入惰性气体,逐步升温至310-320℃并冷凝回流20-40min,冷却至室温后加入乙醇沉淀,取该沉淀溶于正己烷或环己烷中,离心取清液,再向清液中加入适量乙醇,离心取沉淀,如此重复洗涤3次,得到的黑色固体Fe3O4纳米晶,溶于正己烷或环己烷中,得到Fe3O4分散液;
(3)将油酸钠溶于80-100℃的去离子水中,取步骤(2)配好的Fe3O4分散液加至油酸钠溶液中,超声分散5-20min后,放入烘箱中40-70℃干燥至溶液不分层,得到混合物A;
(4)以2,4-二羟基苯甲酸、六次甲基四胺、步骤(3)得到的混合物A为原料,溶于80-100℃的去离子水中,超声分散5-20min后,在160℃下保温4h,在弱酸-弱碱相互作用的诱导下,通过水热过程分子进行自组装,生成具有空腔的、内部包含有Fe3O4纳米晶的空心聚合物壳纳米球(hollowpolymernanospheres,简称h-P),即Fe3O4@h-P,自然冷却后离心、洗涤、干燥成Fe3O4@h-P粉末;
(5)采用热解法,在弱还原气氛中(H2/Ar=5%/95%),在500℃下加热2h,使Fe3O4@h-P的聚合物壳碳化,生成具有空腔的、内部包含有含有Fe3O4纳米晶的空心碳壳纳米球(hollow carbon shell nanospheres,简称h-C),即Fe3O4@h-C;
(6)采用溶剂挥发法,以上转换发光纳米粒子(upconversion nanoparticles,简称UCNPs)的环己烷溶液与步骤(5)中合成的Fe3O4@h-C为原料,先将Fe3O4@h-C溶于乙醇中,再将UCNPs的环己烷溶液用环己烷稀释,其后将Fe3O4@h-C的乙醇溶液在搅拌下缓慢加入该混合溶液中,搅拌20-50min后,加热至40-60℃,敞口搅拌至溶剂除尽,UCNPs均匀附着至Fe3O4@h-C上,冷却后将所得固体用环己烷反复离心洗涤,至离心所得上清液用980nm激光照射观察不到荧光,取离心所得固体,既为Fe3O4@h-C@UCNPs;
(7)采用EDC偶联法合成C18PMH-mPEG,将聚(马来酸酐-1-十八碳烯)(C18PMH)与溶于CH2Cl2/TEA混合溶液中的氨基聚乙二醇(mPEG-NH2)在室温下反应2-4h,然后加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl)为偶联剂,再搅拌12-36h,得到其mPEG-C18PMH粗产物,采用超纯水对所得mPEG-C18PMH粗产物进行透析,通过透析膜(截留分子量MWCO为8000-14000)两天,最后冷冻干燥,即得C18PMH-mPEG粉末;所述C18PMH与mPEG-NH2质量比为1:5;
(8)通过油酸链-C18链的疏水作用,在Fe3O4@h-C@UCNPs表面负载mPEG-C18PMH,先将步骤(6)所得Fe3O4@h-C@UCNPs分散于氯仿中,得到Fe3O4@h-C@UCNPs的氯仿分散液,再将步骤(7)所得C18PMH-mPEG溶于等体积的氯仿中,其后缓慢加入Fe3O4@h-C@UCNPs的氯仿分散液,升温至30-45℃并进行敞口搅拌,直至氯仿挥发完全,得到灰黑色胶状物,立即加入去离子水,继续搅拌使固体均匀分散,离心后用去离子水反复洗涤,既得具有亲水性的Fe3O4@h-C@UCNPs@PEG。
其中,所述UCNPs包括有NaGdF4:Yb3+/Er3+、NaGdF4:Yb3+/Er3+@NaGdF4、NaGdF4:Yb3+/Tm3+、NaGdF4:Yb3+/Tm3+@NaGdF4。
进一步地,所述步骤(1)中油酸钠与氯化铁的摩尔比为1:1。
进一步地,所述步骤(2)中油酸铁、油酸、十八烯的摩尔比为:2:1:39.6。
进一步地,所述步骤(3)中油酸钠与Fe3O4纳米晶的质量比为20:3。
进一步地,所述步骤(6)中UCNPs与Fe3O4@h-C的质量比为1:170~1:50。
进一步地,所述步骤(7)中C18PMH与mPEG-NH2质量比为1:5。
进一步地,所述步骤(8)中mPEG-C18PMH与Fe3O4@h-C@UCNPs的质量比为1:2~5:6。
一种用于肿瘤成像与治疗的纳米材料,该纳米材料具有核-壳结构,包括外部壳层与内部核心,所述外部壳层包括碳化聚合物壳层,碳化聚合物壳层上还附着有上转换发光纳米粒子(UCNPs)层,上转换发光纳米粒子层表面经过PEG改性修饰,所述内部核心为均匀分散在内部空腔中的具有超顺磁性的Fe3O4纳米晶,所述纳米材料具有磁靶向、磁共振成像(MRI)、上转换发光成像、光热治疗功能,可实现肿瘤的诊断治疗一体化。
其中,所述上转换发光纳米粒子层由若干上转换发光纳米粒子均匀排列而成,该上转换发光纳米粒子包括有NaGdF4:Yb3+/Er3+、NaGdF4:Yb3+/Er3+@NaGdF4、NaGdF4:Yb3+/Tm3+、NaGdF4:Yb3+/Tm3+@NaGdF4。
本发明的有益效果为:
(1)用于肿瘤成像与治疗的纳米材料表面经过双亲性聚合物PEG改性修饰,具有良好的水溶性;
(2)用于肿瘤成像与治疗的纳米材料同时具有磁靶向、磁共振成像(MRI)、上转换发光成像、光热治疗等功能,可实现肿瘤的诊断治疗一体化;
(3)用于肿瘤成像与治疗的纳米材料本身生物相容性好,治疗时的毒副作用较小,磁性强度可控;
(4)用于肿瘤成像与治疗的纳米材料的制备方法重复性高,操作易实现,合成过程对设备要求较低,有望应用于生物医学领域。
上述是发明技术方案的概述,以下结合附图与具体实施方式,对本发明做进一步说明。
附图说明
图1为Fe3O4纳米晶(A),自组装后的Fe3O4@h-C(B),Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4(C)及改性后D的Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG(D)的透射电镜图;
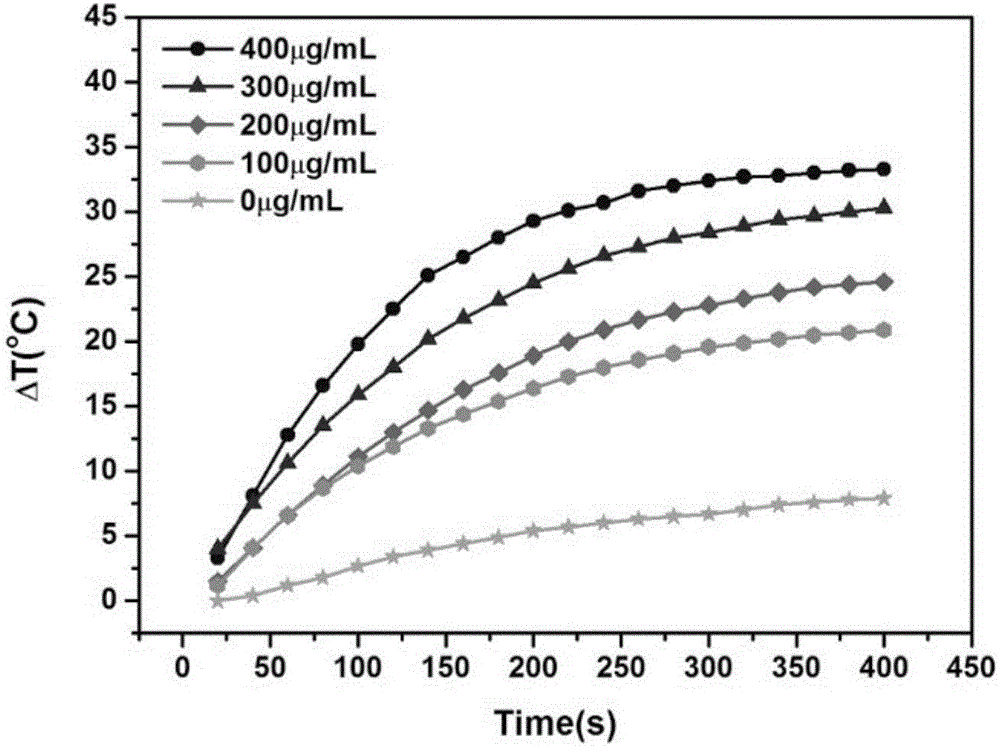
图2为不同浓度Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG在功率密度为1.5W/cm2的808nm近红外光照射0-400s的光热转换曲线;
图3为不同浓度Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG在功率密度为160W/cm2的980nm激光照射下的上转换荧光光谱图;
图4为不同浓度Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG的3T磁共振成像图。
具体实施方式
为更进一步阐述本发明为达到预定目的所采取的技术手段及功效,以下结合附图及较佳实施例,对本发明的具体实施方式详细说明。
实施例1
本发明实施例提供一种用于肿瘤成像与治疗的纳米材料及其制备方法,包括以下步骤:
(1)将FeCl3·6H2O和油酸钠(摩尔比为1:1)加至乙醇、去离子水与正己烷的混合液(体积比为4:3:7)中,超声溶解完全后,在PTFE搅拌桨搅拌下,70℃冷凝回流,油浴加热4h,得到红棕色粘稠液,将其转移至梨形分液漏斗中,加入适量去离子水,振荡,静置,弃去水相,重复洗涤3次,取油相于110℃下真空干燥一夜,得油酸铁;
(2)采用热裂解法,取步骤(1)制得的油酸铁、油酸和十八烯(摩尔比为2:1:39.6)于三口烧瓶中,超声分散完全后,在真空条件下,110℃加热30min。在惰性气体保护下,以3℃min-1的速度升温,将上述混合物加热至320℃,冷凝回流维持30min,得棕黑色浑浊液,在惰性气体保护下冷却至室温。向棕黑色浑浊液中加入5-10倍体积的乙醇,离心取沉淀,然后将沉淀溶于适量正己烷,离心取清液,再向清液中加入适量乙醇,离心取固体,如此重复洗涤3次,得到的黑色固体Fe3O4纳米晶,溶于正己烷中,配成30mg/mL的Fe3O4分散液密封储存;
(3)将油酸钠溶于80℃去离子水,取步骤(2)配好的Fe3O4的正己烷分散液加至油酸钠溶液中,超声分散10min后,放入烘箱中50℃干燥至溶液不分层,得混合物A;其中,油酸钠与Fe3O4纳米晶的质量比为20:3;
(4)将2,4-二羟基苯甲酸和六次甲基四胺(摩尔比4:1)加至80℃去离子水中,溶解完全后,加入步骤(3)得到的混合物A,超声分散10min,其后转移至反应釜中,放入烘箱160℃维持4h后自然冷却,离心取固体。再用去离子水离心洗涤多次,最后用乙醇离心洗涤1次。将离心得到的固体于烘箱中50℃干燥一夜,得到Fe3O4@h-P粉末,置于干燥器储存;
(5)采用热解法,采用热解法,在弱还原气氛中(H2/Ar=5%/95%),在500℃下加热2h,使Fe3O4@h-P的聚合物壳碳化,生成具有空腔的、内部包含有含有Fe3O4纳米晶的空心碳壳纳米球,即Fe3O4@h-C;
(6)采用溶剂热法,以GdCl3·6H2O、YbCL3·6H2O和ErCl3·6H2O(摩尔比56:10:1)为原料,分散于油酸/1-十八碳烯的混合溶液中,在150℃下搅拌1h后,加入NaOH和NH4F的甲醇溶液,在300℃与氩气保护下搅拌1h,反应完成后冷却,加入丙酮中进行离心获得NaGdF4:Yb3+/Er3+,并用环己烷和丙酮洗涤两次,制得NaGdF4:Yb3+/Er3+,并将其分散于环己烷溶液中,得到NaGdF4:Yb3+/Er3+的环己烷溶液;
(7)采用溶剂挥发法,以NaGdF4:Yb3+/Er3+的环己烷溶液与步骤(5)中合成的Fe3O4@h-C为原料(NaGdF4:Yb3+/Er3+与Fe3O4@h-C质量比1:170),先将Fe3O4@h-C溶于乙醇中,密封磁力搅拌一夜,再将NaGdF4:Yb3+/Er3+的环己烷溶液用环己烷稀释,其后将Fe3O4@h-C的乙醇溶液在搅拌下缓慢加入该混合溶液中,搅拌30min后,加热至50℃,敞口搅拌至溶剂除尽,NaGdF4:Yb3+/Er3+逐渐附着至Fe3O4@h-C上,冷却后将所得固体用环己烷反复离心洗涤,至离心所得上清液用980nm激光照射观察不到荧光,取离心所得固体,既为Fe3O4@h-C@NaGdF4:Yb3+/Er3+;
(8)采用EDC偶联法合成mPEG-C18PMH,将聚(马来酸酐-1-十八碳烯)(C18PMH)与溶于CH2Cl2/TEA混合溶液中的氨基聚乙二醇(mPEG-NH2)在室温下反应2h,然后加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl)为偶联剂,再搅拌24h,得到其后C18PMH-mPEG粗产物,采用超纯水对所得mPEG-C18PMH粗产物进行透析,通过透析膜(截留分子量MWCO为8000-14000)两天,最后冷冻干燥,即得mPEG-C18PMH粉末;所述C18PMH与mPEG-NH2质量比为1:5;
(9)通过油酸链-C18链的疏水作用,在Fe3O4@h-C@NaGdF4:Yb3+/Er3+表面负载mPEG-C18PMH,首先将步骤(7)得到的Fe3O4@h-C@NaGdF4:Yb3+/Er3+分散于等体积的氯仿中,得到Fe3O4@h-C@NaGdF4:Yb3+/Er3+的氯仿分散液,将步骤(8)所得的mPEG-C18PMH溶于等体积的氯仿中,其后缓慢加入Fe3O4@h-C@NaGdF4:Yb3+/Er3+的氯仿分散液,升温至30℃敞口搅拌,直至氯仿挥发完全,得到灰黑色胶状物。然后,立刻加入适量去离子水,继续搅拌,使固体均匀分散,其后用去离子水离心洗涤固体多次,既得Fe3O4@h-C@NaGdF4:Yb3+/Er3+@PEG,最后将离心所得固体溶于去离子水中,4℃密封保存,其中C18PMH-mPEG与Fe3O4@h-C@NaGdF4:Yb3+/Er3+的质量比为1:2。
实施例2
本发明实施例提供另一用于肿瘤成像与治疗的纳米材料及其制备方法,包括以下步骤:
(1)将FeCl3·6H2O和油酸钠(摩尔比为1:1)加至乙醇、去离子水与环己烷的混合液(体积比为4:3:7)中,超声溶解完全后,在PTFE搅拌桨搅拌下,60℃冷凝回流,油浴加热6h,得到红棕色粘稠液,将其转移至梨形分液漏斗中,加入适量去离子水,振荡,静置,弃去水相,重复洗涤3次,取油相于140℃下真空干燥一夜,得油酸铁;
(2)采用热裂解法,取步骤(1)制得的油酸铁、油酸和十八烯(摩尔比为2:1:39.6)于三口烧瓶中,超声分散完全后,在真空条件下,100℃加热40min。在惰性气体保护下,以4℃min-1的速度升温,将上述混合物加热至350℃,冷凝回流维持20min,得棕黑色浑浊液,在惰性气体保护下冷却至室温。向棕黑色浑浊液中加入5-10倍体积的乙醇,离心取沉淀,然后将沉淀溶于适量环己烷,离心取清液,再向清液中加入适量乙醇,离心取固体,如此重复洗涤3次,得到的黑色固体Fe3O4纳米晶,溶于环己烷中,配成30mg/mL的Fe3O4分散液密封储存;
(3)将油酸钠溶于90℃去离子水,取步骤(2)配好的Fe3O4的环己烷分散液加至油酸钠溶液中,超声分散20min后,放入烘箱中40℃干燥至溶液不分层,得混合物A;其中,油酸钠与Fe3O4纳米晶的质量比为20:3;
(4)将2,4-二羟基苯甲酸和六次甲基四胺(摩尔比4:1)加至90℃去离子水中,溶解完全后,加入步骤(3)得到的混合物A,超声分散20min,其后转移至反应釜中,放入烘箱160℃维持4h后自然冷却,离心取固体。再用去离子水离心洗涤多次,最后用乙醇离心洗涤1次。将离心得到的固体于烘箱中50℃干燥一夜,得到Fe3O4@h-P粉末,置于干燥器储存;
(5)采用热解法,采用热解法,在弱还原气氛中(H2/Ar=5%/95%),在500℃下加热2h,使Fe3O4@h-P的聚合物壳碳化,生成具有空腔的、内部包含有含有Fe3O4纳米晶的空心碳壳纳米球,即Fe3O4@h-C;
(6)采用溶剂热法,以GdCl3·6H2O、YbCL3·6H2O和ErCl3·6H2O(摩尔比56:10:1)为原料,分散于油酸/1-十八碳烯的混合溶液中,在150℃下搅拌1h后,加入NaOH和NH4F的甲醇溶液,在300℃与氩气保护下搅拌1h,反应完成后冷却,加入丙酮中进行离心获得NaGdF4:Yb3+/Er3+,并用环己烷和丙酮洗涤两次,制得NaGdF4:Yb3+/Er3+,并将其分散于环己烷溶液中,得到NaGdF4:Yb3+/Er3+的环己烷溶液;
(7)将GdCl3加入到三颈圆底烧瓶中,并加热至110℃以蒸发除去水。然后在150℃下加入油酸和十八烷的复合溶液,待GdCl3完全溶解后冷却至60℃。接着,加入NaGdF4:Yb3+/Er3+、NaOH和NH4F的甲醇溶液,将溶液在110℃加热30分钟,以除去甲醇,环己烷。将所得溶液在300℃氩气下反应1小时。反应完成后,通过在加入丙酮中离心获得NaGdF4:Yb3+/Er3+@NaGdF4的粗产品,并用环己烷和丙酮洗涤两次后,再分散在环己烷中,得到NaGdF4:Yb3+/Er3+@NaGdF4的环己烷溶液。
(8)采用溶剂挥发法,以NaGdF4:Yb3+/Er3+@NaGdF4的环己烷溶液与步骤(5)中合成的Fe3O4@h-C为原料(NaGdF4:Yb3+/Er3+@NaGdF4与Fe3O4@h-C质量比1:50),先将Fe3O4@h-C溶于乙醇中,密封磁力搅拌一夜,再将NaGdF4:Yb3+/Er3+@NaGdF4的环己烷溶液用环己烷稀释,其后将Fe3O4@h-C的乙醇溶液在搅拌下缓慢加入该混合溶液中,搅拌30min后,加热至40℃,敞口搅拌至溶剂除尽,NaGdF4:Yb3+/Er3+@NaGdF4逐渐附着至Fe3O4@h-C上,冷却后将所得固体用环己烷反复离心洗涤,至离心所得上清液用980nm激光照射观察不到荧光,取离心所得固体,既为Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4;
(9)采用EDC偶联法合成mPEG-C18PMH,将聚(马来酸酐-1-十八碳烯)(C18PMH)与溶于CH2Cl2/TEA混合溶液中的氨基聚乙二醇(mPEG-NH2)在室温下反应3h,然后加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl)为偶联剂,再搅拌12h,得到其后mPEG-C18PMH粗产物,采用超纯水对所得mPEG-C18PMH粗产物进行透析,通过透析膜(截留分子量MWCO为8000-14000)两天,最后冷冻干燥,即得mPEG-C18PMH粉末;所述C18PMH与mPEG-NH2质量比为1:5;
(10)通过油酸链-C18链的疏水作用,在Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4表面负载mPEG-C18PMH,首先将步骤(7)得到的Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4分散于等体积的氯仿中,得到Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4的氯仿分散液,将步骤(8)所得的mPEG-C18PMH溶于等体积的氯仿中,其后缓慢加入Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4的氯仿分散液,升温至45℃敞口搅拌,直至氯仿挥发完全,得到灰黑色胶状物。然后,立刻加入适量去离子水,继续搅拌,使固体均匀分散,其后用用去离子水离心洗涤固体多次,既得Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG最后将离心所得固体溶于去离子水中,4℃密封保存,其中mPEG-C18PMH与Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4的质量比为5:6。
图1为Fe3O4(A),Fe3O4@h-C(B),Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4(C)和Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG(D)的透射电镜图。从A图中可以看出,Fe3O4大小形貌均一,分散性好,由粒度分析软件nano measurer1.2.5统计100个纳米粒子得出Fe3O4的粒径为6.44±1.14nm。B图显示Fe3O4@h-C形状规则,球体内部存在较大空腔,空腔内包含有Fe3O4纳米晶,球体表面较光滑,统计40个纳米粒子得Fe3O4@h-C的粒径为334.35±30.23nm,壳层厚度的平均值为61.40nm。C图显示Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4为表面均匀附着NaGdF4:Yb3+/Er3+@NaGdF4的规则的纳米球体。D图显示改性后的Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG纳米球表面,NaGdF4:Yb3+/Er3+@NaGdF4能稳定附着。
称取Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG,配成400μg/mL的水分散液,并用去离子水稀释,配成300μg/mL,200μg/mL,100μg/mL的具有浓度梯度的水分散液。取等体积的各浓度的分散液,于同规格的试剂瓶中。在波长为808nm,功率密度为1.5W/cm2的近红外光的激发下,每隔20s记录各试剂瓶中分散液的温度,当激光照射至400s时,关闭激光。温度与时间的关系如图2所示。
图2为不同浓度Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG在功率密度为1.5W/cm2的808nm近红外光照射0-400s的光热转换曲线。图2的数据表明,材料的浓度越高,升温的速度越快,并且最终的温度也最高。在约300s后,各个浓度的材料温度都维持在一个稳定的值。
称取Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG,配成400μg/mL的水分散液,并用去离子水稀释,配成300μg/mL,200μg/mL,100μg/mL的具有浓度梯度的水分散液。取Gd3+的理论浓度为0.47M的NaGdF4:Yb3+/Er3+@NaGdF4环己烷溶液100μL,用环己烷稀释至2mL。
取上述配好的样品2mL置于1.0cm×1.0cm石英比色皿中,在功率密度为160W/cm2的980nm激光照射下测上转换荧光光谱,如图3所示。
图3为实施例1制备的不同浓度Fe3O4@h-C@UCNPs@PEG在功率密度为160W/cm2的980nm激光照射下的上转换荧光光谱图。图3表明粒子浓度越高,荧光强度越大。
将Fe3O4@h-C@NaGdF4:Yb3+/Er3+@NaGdF4@PEG通过ICP-AES测试法测定分散液中Gd元素的浓度。然后用去离子水配置Gd浓度依次为0.2、0.1、0.05、0.025、0.0125mM的分散液,通过磁共振成像测定不同Gd浓度下的T1弛豫效应如图4所示。
图4实施例1制备的不同浓度Fe3O4@h-C@NaGdF4:Yb3+/Er3+@PEG的3T磁共振成像图。图4显示样品浓度增加,样品的影像越明亮,说明本材料具有良好的T1造影效果。
实施例3
本发明实施例提供另一用于肿瘤成像与治疗的纳米材料及其制备方法,包括以下步骤:
(1)将FeCl3·6H2O和油酸钠(摩尔比为1:1)加至乙醇、去离子水与正己烷的混合液(体积比为4:3:7)中,超声溶解完全后,在磁力搅拌下,80℃冷凝回流,油浴加热3h,得到红棕色粘稠液,将其转移至梨形分液漏斗中,加入适量去离子水,振荡,静置,弃去水相,重复洗涤3次,取油相于100℃下真空干燥一夜,得油酸铁;
(2)采用热裂解法,取步骤(1)制得的油酸铁、油酸和十八烯(摩尔比为2:1:39.6)于三口烧瓶中,超声分散完全后,在真空条件下,120℃加热20min。在惰性气体保护下,以3℃min-1的速度升温,将上述混合物加热至310℃,冷凝回流维持40min,得棕黑色浑浊液,在惰性气体保护下冷却至室温。向棕黑色浑浊液中加入5-10倍体积的乙醇,离心取沉淀,然后将沉淀溶于适量正己烷,离心取清液,再向清液中加入适量乙醇,离心取固体,如此重复洗涤3次,得到的黑色固体Fe3O4纳米晶,溶于正己烷中,配成30mg/mL的Fe3O4分散液密封储存;
(3)将油酸钠溶于100℃去离子水,取步骤(2)配好的Fe3O4的正己烷分散液加至油酸钠溶液中,超声分散5min后,放入烘箱中70℃干燥至溶液不分层,得混合物A;其中,油酸钠与Fe3O4纳米晶的质量比为20:3;
(4)将2,4-二羟基苯甲酸和六次甲基四胺(摩尔比4:1)加至100℃去离子水中,溶解完全后,加入步骤(3)得到的混合物A,超声分散5min,其后转移至反应釜中,放入烘箱160℃维持4h后自然冷却,离心取固体。再用去离子水离心洗涤多次,最后用乙醇离心洗涤1次。将离心得到的固体于烘箱中50℃干燥一夜,得到Fe3O4@h-P粉末,置于干燥器储存;
(5)采用热解法,采用热解法,在弱还原气氛中(H2/Ar=5%/95%),在400℃下加热3h,使Fe3O4@h-P的聚合物壳碳化,生成具有空腔的、内部包含有含有Fe3O4纳米晶的空心碳壳纳米球,即Fe3O4@h-C;
(6)采用溶剂热法,以Gd(CH3COO)3、Tm(CH3COO)3和Yb(CH3COO)3(摩尔比49:49:2)为原料,分散于油酸/十八烷的混合溶液中,在140℃下搅拌3h后,加入NaOH和NH4F的甲醇溶液,在200℃与氩气保护下搅拌3h,反应完成后冷却,加入丙酮中进行离心获得NaGdF4:Yb3+/Tm3+,并用环己烷和丙酮洗涤两次,制得NaGdF4:Yb3+/Tm3+,并将其分散于环己烷溶液中,得到NaGdF4:Yb3+/Tm3+的环己烷溶液;
(7)采用溶剂挥发法,以NaGdF4:Yb3+/Tm3+的环己烷溶液与步骤(5)中合成的Fe3O4@h-C为原料(NaGdF4:Yb3+/Tm3+与Fe3O4@h-C质量比1:170),先将Fe3O4@h-C溶于乙醇中,密封磁力搅拌一夜,再将NaGdF4:Yb3+/Tm3+的环己烷溶液用环己烷稀释,其后将Fe3O4@h-C的乙醇溶液在搅拌下缓慢加入该混合溶液中,搅拌30min后,加热至60℃,敞口搅拌至溶剂除尽,NaGdF4:Yb3+/Tm3+逐渐附着至Fe3O4@h-C上,冷却后将所得固体用环己烷反复离心洗涤,至离心所得上清液用980nm激光照射观察不到荧光,取离心所得固体,既为Fe3O4@h-C@NaGdF4:Yb3+/Tm3+;
(8)采用EDC偶联法合成mPEG-C18PMH,将聚(马来酸酐-1-十八碳烯)(C18PMH)与溶于CH2Cl2/TEA混合溶液中的氨基聚乙二醇(mPEG-NH2)在室温下反应4h,然后加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl)为偶联剂,再搅拌36h,得到其后C18PMH-mPEG粗产物,采用超纯水对所得mPEG-C18PMH粗产物进行透析,通过透析膜(截留分子量MWCO为14000)两天,最后在常温真空下冷却干燥,即得mPEG-C18PMH粉末;所述C18PMH与mPEG-NH2质量比为1:5;
(9)通过油酸链-C18链的疏水作用,在Fe3O4@h-C@NaGdF4:Yb3+/Tm3+表面负载mPEG-C18PMH,首先将步骤(7)得到的Fe3O4@h-C@NaGdF4:Yb3+/Tm3+分散于等体积的氯仿中,得到Fe3O4@h-C@NaGdF4:Yb3+/Tm3+的氯仿分散液,将步骤(8)所得的mPEG-C18PMH溶于等体积的氯仿中,其后缓慢加入Fe3O4@h-C@NaGdF4:Yb3+/Tm3+的氯仿分散液,升温至40℃敞口搅拌,直至氯仿挥发完全,得到灰黑色胶状物。然后,立刻加入适量去离子水,继续搅拌,使固体均匀分散,其后用用去离子水离心洗涤固体多次,既得Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@PEG,最后将离心所得固体溶于去离子水中,4℃密封保存,其中mPEG-C18PMH与Fe3O4@h-C@NaGdF4:Yb3+/Tm3+的质量比为1:2。
实施例4
本发明实施例提供另一用于肿瘤成像与治疗的纳米材料及其制备方法,包括以下步骤:
(1)将FeCl3·6H2O和油酸钠(摩尔比为1:1)加至乙醇、去离子水与正己烷的混合液(体积比为4:3:7)中,超声溶解完全后,在磁力搅拌下,70℃冷凝回流,油浴加热4h,得到红棕色粘稠液,将其转移至梨形分液漏斗中,加入适量去离子水,振荡,静置,弃去水相,重复洗涤3次,取油相于130℃下真空干燥一夜,得油酸铁;
(2)采用热裂解法,取步骤(1)制得的油酸铁、油酸和十八烯(摩尔比为2:1:39.6)于三口烧瓶中,超声分散完全后,在真空条件下,110℃加热30min。在惰性气体保护下,以4℃min-1的速度升温,将上述混合物加热至330℃,冷凝回流维持30min,得棕黑色浑浊液,在惰性气体保护下冷却至室温。向棕黑色浑浊液中加入5-10倍体积的乙醇,离心取沉淀,然后将沉淀溶于适量正己烷,离心取清液,再向清液中加入适量乙醇,离心取固体,如此重复洗涤3次,得到的黑色固体Fe3O4纳米晶,溶于正己烷中,配成30mg/mL的Fe3O4分散液密封储存;
(3)将油酸钠溶于85℃去离子水,取步骤(2)配好的Fe3O4的正己烷分散液加至油酸钠溶液中,超声分散10min后,放入烘箱中60℃干燥至溶液不分层,得混合物A;其中,油酸钠与Fe3O4纳米晶的质量比为20:3;
(4)将2,4-二羟基苯甲酸和六次甲基四胺(摩尔比4:1)加至90℃去离子水中,溶解完全后,加入步骤(3)得到的混合物A,超声分散10min,其后转移至反应釜中,放入烘箱160℃维持4h后自然冷却,离心取固体。再用去离子水离心洗涤多次,最后用乙醇离心洗涤1次。将离心得到的固体于烘箱中50℃干燥一夜,得到Fe3O4@h-P粉末,置于干燥器储存;
(5)采用热解法,采用热解法,在弱还原气氛中(H2/Ar=5%/95%),在500℃下加热2h,使Fe3O4@h-P的聚合物壳碳化,生成具有空腔的、内部包含有含有Fe3O4纳米晶的空心碳壳纳米球,即Fe3O4@h-C;
(6)采用溶剂热法,以Gd(CH3COO)3、Tm(CH3COO)3和Yb(CH3COO)3(摩尔比49:49:2)为原料,分散于油酸/十八烷的混合溶液中,在140℃下搅拌1h后,加入NaOH和NH4F的甲醇溶液,在300℃与氩气保护下搅拌1h,反应完成后冷却,加入丙酮中进行离心获得NaGdF4:Yb3+/Tm3+,并用环己烷和丙酮洗涤两次,制得NaGdF4:Yb3+/Tm3+,并将其分散于环己烷溶液中,得到NaGdF4:Yb3+/Tm3+的环己烷溶液;
(7)将GdCl3加入到三颈圆底烧瓶中,并加热至110℃以蒸发除去水。然后在150℃下加入油酸和十八烷的复合溶液,待GdCl3完全溶解后冷却至60℃。接着,加入NaGdF4:Yb3+/Tm3+、NaOH和NH4F的甲醇溶液,将溶液在100℃加热30分钟,以除去甲醇,环己烷。将所得溶液在200℃氩气下反应2小时。反应完成后,通过在加入丙酮中离心获得NaGdF4:Yb3+/Tm3+@NaGdF4的粗产品,并用环己烷和丙酮洗涤两次后,再分散在环己烷中,得到NaGdF4:Yb3+/Tm3+@NaGdF4的环己烷溶液。
(8)采用溶剂挥发法,以NaGdF4:Yb3+/Tm3+@NaGdF4的环己烷溶液与步骤(5)中合成的Fe3O4@h-C为原料(NaGdF4:Yb3+/Tm3+@NaGdF4与Fe3O4@h-C质量比1:50),先将Fe3O4@h-C溶于乙醇中,密封磁力搅拌一夜,再将NaGdF4:Yb3+/Tm3+@NaGdF4的环己烷溶液用环己烷稀释,其后将Fe3O4@h-C的乙醇溶液在搅拌下缓慢加入该混合溶液中,搅拌30min后,加热至50℃,敞口搅拌至溶剂除尽,NaGdF4:Yb3+/Tm3+@NaGdF4逐渐附着至Fe3O4@h-C上,冷却后将所得固体用环己烷反复离心洗涤,至离心所得上清液用980nm激光照射观察不到荧光,取离心所得固体,既为Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4;
(9)采用EDC偶联法合成mPEG-C18PMH,将聚(马来酸酐-1-十八碳烯)(C18PMH)与溶于CH2Cl2/TEA混合溶液中的氨基聚乙二醇(mPEG-NH2)在室温下反应2h,然后加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC·HCl)为偶联剂,再搅拌24h,得到其后mPEG-C18PMH粗产物,采用超纯水对所得mPEG-C18PMH粗产物进行透析,通过透析膜(截留分子量MWCO为8000-14000)两天,最后在常温真空下冷却干燥,即得mPEG-C18PMH粉末;所述C18PMH与mPEG-NH2质量比为1:5;
(10)通过油酸链-C18链的疏水作用,在Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4表面负载mPEG-C18PMH,首先将步骤(7)得到的Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4分散于等体积的氯仿中,得到Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4的氯仿分散液,将步骤(8)所得的mPEG-C18PMH溶于等体积的氯仿中,其后缓慢加入Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4的氯仿分散液,升温至35℃敞口搅拌,直至氯仿挥发完全,得到灰黑色胶状物。然后,立刻加入适量去离子水,继续搅拌,使固体均匀分散,其后用用去离子水离心洗涤固体多次,既得Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4@PEG,最后将离心所得固体溶于去离子水中,4℃密封保存,其中mPEG-C18PMH与Fe3O4@h-C@NaGdF4:Yb3+/Tm3+@NaGdF4的质量比为5:6。
本发明的重点主要在于,用于肿瘤成像与治疗的纳米材料表面经过双亲性聚合物PEG改性修饰,具有良好的水溶性;同时具有磁靶向、磁共振成像(MRI)、上转换发光成像、光热治疗等功能,可实现肿瘤的诊断治疗一体化;纳米材料本身生物相容性好,治疗时的毒副作用较小,磁性强度可控;制备方法重复性高,操作易实现,合成过程对设备要求较低,有望应用于生物医学领域。
以上所述,仅是本发明的较佳实施例而已,并非对本发明的技术范围作任何限制,故采用与本发明上述实施例相同或近似的技术特征,均在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 酸敏感可控释放单线态氧纳米材料及其制备方法与应用与流程
- 一种层状复合纳米材料M‑LDHs@Si02微球的制备方法与流程
- 复合纳米材料和微米材料、它们的膜和其制备方法及用途与流程
- 复合纳米材料和微米材料、它们的膜和其制备方法及用途与流程
- 包括离散氧化铝纳米材料的正电极以及形成氧化铝纳米材料的方法与流程
- 一种基于TiO2介晶纳米材料的双响应夹心型免疫传感器的制备方法及应用与流程
- 一种基于核酸和铂纳米材料的自组装体系检测核酸浓度的方法与流程
- 一种高容量蜂窝结构的ZnCo2S4纳米材料及其制备和应用的制作方法与工艺
- 一种银纳米棒/聚合物/银纳米片核壳纳米材料及其制备方法和应用与流程
- 一种WC‑SiO2‑Mn‑Fe纳米材料及其制备方法与流程