固体分散体及其制备方法和应用与流程
本发明涉及医药技术领域,特别是涉及一种固体分散体及其制备方法和应用。
背景技术:
口服给药是最简单最容易的给药途径。由于口服固体制剂和其他口服剂型相比具有独特优势,如理化性质稳定性好,生产工艺简单,服用携带方便,剂量准确。口服固体制剂是大部分正在开发新化学实体的首选药物传递方式。但要求口服后能被有效吸收,产生重现性良好的血药浓度。由于药物在体内必须先溶解才能透过生物膜,被吸收进入血液循环,才能获得理想的生物利用度。有调查表明,BCSⅡ类药物在上市药物中的比例为30%,而这一比例在在研药物中更是达到了70%。因此,提高难溶性药物的溶出速率以提高药物的生物利用度已经成为药学研究的热点和难点。
尽管成盐、增溶、微粉化等技术经常被用于提高难溶性药物的溶出速率,从而增加其口服吸收和生物利用度,但这些方法均具有一定的局限性,商业化应用受到限制。成盐法不能用于中性药物,并且对于弱酸或弱碱性药物来说,要选择合适的成盐法较困难。即使能够成盐,很多情况下这些盐在胃肠道环境不稳定,易转化为各自的酸或碱的形式,对溶解度的提高有限。利用表面活性剂或助溶剂可以增加药物在有机溶剂或水中的溶解度并进而制成液体制剂,但这势必会降低患者的顺应性以及产品的市场前景。微粉化技术对粒径减少的限度有限,后续制剂处理困难,粉末疏水性增加并有可能再聚集使粉末的润湿性降低。而多晶型和溶剂化物在溶解时可能会从亚稳定态转变为稳定态。
固体分散技术的开发和应用为难溶性药物的生物利用度问题提供了一种很好的解决方法。固体分散技术是指将药物以分子、无定形或微晶状态高度均匀分散在固体载体中的技术。自1961年固体分散技术首次用于提高难溶性药物的溶出速率和口服生物利用度以来,诸多研究者对固体分散体进行了广泛深入的研究,进一步证明将难溶性药物制成固体分散体是提高其溶解度和溶出速率的最具应用潜力的方法之一。固体分散体提高难溶性药物溶出的机制主要包括:减小药物粒径、增加润湿性、改变药物物理状态(将药物由结晶变为无定形或分子状态分散在载体中)。其具体机制可能是上述一种或几种机制共同作用的结果。近年来,人们运用固体分散技术,采用不同性质的载体材料使药物在高度分散状态下达到不同的用药目的。利用水溶性高分子载体材料制备固体分散体,能够增加难溶性药物的溶解度和溶出速率,从而提高药物的生物利用度;利用水不溶性或脂溶性载体材料制备固体分散体,则不但能够提高药物的生物利用度,还能延缓或控制药物的释放;利用肠溶性载体,可控制药物在肠道释放;其次,利用载体材料的包蔽作用,可延缓或阻止药物的水解和氧化,提高药物在贮存期的化学稳定性,掩盖药物的不良臭味和刺激性。固体分散体作为一种中间体,还可根据需要制备成制成胶囊剂、片剂、微丸剂、栓剂、滴丸等多种剂型。固体分散技术在提高难溶性药物的溶出度及生物利用度,制备缓控释制剂以及剂型开发方面具有显著的优越性。
固体分散体的传统制备方法包括熔融法,溶剂法,溶剂-熔融法。近年来,热熔挤出法和喷雾干燥作为新型的制备固体分散体的方法备受国内外药剂学研究者的关注。以上制备方法除了喷雾干燥法,其他方法均需药物与载体在熔融状态下进行制备,因此,对于具有较高熔点的药物和载体,容易导致药物和载体的热分解,从而限制了此方法的广泛应用。喷雾干燥具有混合性强、连续操作性好、工艺温度较低等优势,但是,现有技术中喷雾干燥的方法需要先将难溶性药物溶于有机溶剂中,因此具有有机溶剂残留的缺点。
技术实现要素:
基于此,本发明提供了一种喷雾干燥制备固体分散体的方法,该方法通过添加特定种类的表面活性剂和水溶性高分子材料,使制备过程中无须用到有机溶剂,避免了有机溶剂的残留问题。
具体技术方案如下:
一种喷雾干燥固体分散体的制备方法,包括以下步骤:
(1)将难溶性药物、表面活性剂以及水溶性高分子材料混合均匀后加入水中,加热至50~90℃,搅拌至澄清,得含药混合溶液;所述水的用量与其它原料的总重量的配比为1ml:30-60mg,所述其它原料的总重量为所述难溶性药物、表面活性剂以及水溶性高分子材料的总重量;
(2)将所得含药混合溶液进行喷雾干燥,在喷雾干燥过程中,保持含药混合溶液的温度与步骤(1)中所述加热的温度相同;
所述难溶性药物在所述固体分散体中的质量百分比为3~25%;所述表面活性剂与所述水溶性高分子材料的质量比为1:1~10;
所述表面活性剂选自十二烷基硫酸钠、泊洛沙姆、吐温、α-生育酚、琥珀酸酯、聚乙二醇、胆酸钠和聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物中的至少一种;
所述水溶性高分子材料选自聚维酮、共聚维酮、羟丙甲纤维素和聚乙二醇中的至少一种。
在其中一些实施例中,所述难溶性药物选自:二氟尼柳、吡罗昔康、卡马西平、灰黄霉素、酮康唑。
在其中一些实施例中,所述表面活性剂选自十二烷基硫酸钠、泊洛沙姆F68、吐温80、α-生育酚、琥珀酸酯、聚乙二醇、胆酸钠和聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物中的至少一种;所述水溶性高分子材料选自聚维酮K25、共聚维酮VA64、羟丙甲纤维素E5和聚乙二醇6000中的至少一种。
在其中一些实施例中,所述表面活性剂为聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物,所述水溶性高分子材料为聚维酮K25。
在其中一些实施例中,所述难溶性药物的质量百分比为5~20%。
在其中一些实施例中,所述难溶性药物的质量百分比为10~20%。
在其中一些实施例中,所述表面活性剂与所述水溶性高分子材料的质量比为1:1.5~9。
在其中一些实施例中,所述药物为二氟尼柳,所述表面活性剂为聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物,所述水溶性高分子材料为聚维酮K25,所述表面活性剂与所述水溶性高分子材料的质量比为1:1.5~4.5,所述难溶性药物的质量百分比为10~20%。
在其中一些实施例中,所述药物为二氟尼柳,所述表面活性剂为聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物,所述水溶性高分子材料为聚维酮K25,所述表面活性剂与所述水溶性高分子材料的质量比为1:3.5~4.5,所述难溶性药物的质量百分比为15~20%。
在其中一些实施例中,所述水的用量与其它原料的总重量的配比为1ml:30-35mg,所述其它原料的总重量为所述难溶性药物、表面活性剂以及水溶性高分子材料的总重量。
在其中一些实施例中,所述加热的温度为70-85℃。
在其中一些实施例中,所述喷雾干燥的工艺条件为:进风温度100~140℃、出风温度60~90℃、风量0.5~0.9m3/min、雾化压力7~15kPa、液体流速1~3mL/min。
在其中一些实施例中,所述喷雾干燥的工艺条件为:进风温度110~130℃,出风温度68~80℃,风量0.6~0.8m3/min,雾化压力8~10KPa,液体流速1.5~2.5mL/min。
在其中一些实施例中,所述喷雾干燥固体分散体的制备方法还包括以下步骤:将喷雾干燥后的产物在20-30℃的真空干燥箱中干燥20-28小时后过筛,得到二氟尼柳固体分散体粉末。
本发明还提供了一种固体分散体。
具体技术方案如下:
由上述制备方法制备得到的固体分散体。
本发明还提供了上述固体分散体的应用。
具体技术方案如下:
上述的固体分散体在制备难溶性药物的制剂中的应用。所述制剂的剂型可以是片剂、胶囊剂、微丸剂、栓剂、散剂、滴丸或干混悬剂等多种剂型。
本发明的固体分散体及其制备方法和应用具有以下优点和有益效果:
本发明发明人通过大量的创造性实验研究,发现难溶性药物与特定种类、特定比例的表面活性剂和水溶性高分子材料的混合物,在水中于加热状态下即可获得澄清的溶液,无须用到有机溶剂,难溶性药物在溶液中处于分子状态,再将其进行喷雾干燥,可制备得到无定型的固体分散体。本发明的喷雾干燥法制备固体分散体的过程中无须用到有机溶剂,环境友好,成功解决了现有技术中,喷雾干燥法制备固体分散体有机溶剂残留的问题,避免了服用药物时因有机溶剂残留带来的副作用。
本发明制备的固体分散体能使难溶性药物的溶解度增加,溶出速度和溶出度显著提高,从而能提高难溶性药物的生物利用度,降低给药剂量,减少药物不良反应。跟原料药相比,本发明固体分散体的药物溶解度提高了4-12倍。
本发明进一步优选的难溶性药物为熔点高或者熔融即分解的药物,属于优质的氢键给体,而所选的水溶性高分子材料为氢键受体,药物与水溶性高分子材料能形成氢键,可以更好的提高药物的溶出度。
水溶性高分子材料和表面活性剂分别进一步优选为聚维酮和聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物,优选出的水溶性高分子载体材料和表面活性剂能使难溶性药物固体分散体中的药物溶解度进一步增加,溶出速度和溶出度更快,固体分散体的稳定性更好。
附图说明
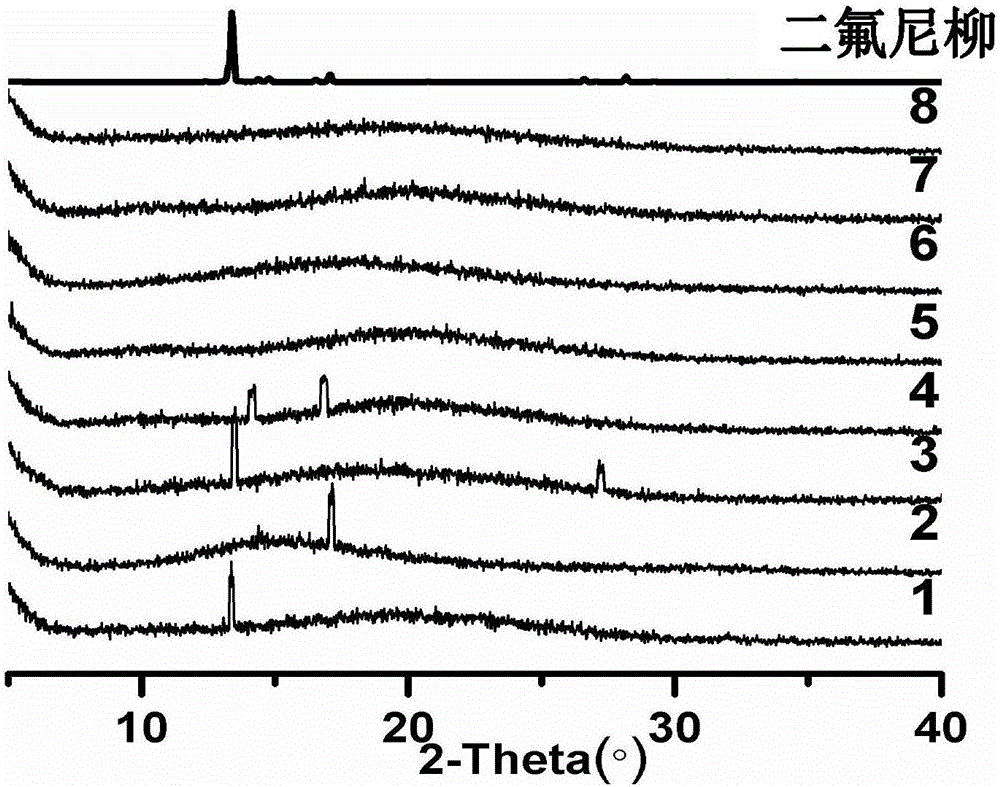
图1为实施例1中编号1-8制备得到的二氟尼柳固体分散体进行X射线衍射测试的图谱;
图2为实施例1中编号1-8制备得到的二氟尼柳固体分散体放置于25℃恒温箱中60天后进行X射线衍射测试的图谱;
图3为为实施例1中编号1-8制备得到的二氟尼柳固体分散体体外溶出曲线;
图4为实施例1中二氟尼柳原料粉末、二氟尼柳固体分散体的体外溶出曲线;
图5为实施例2中吡罗昔康原料粉末、吡罗昔康和聚维酮K25和Soluplus物理混合物粉末、聚维酮K25与Soluplus物理混合物粉末、吡罗昔康固体分散体粉末以及吡罗昔康固体分散体六个月稳定性的X射线衍射图谱;
图6为实施例2中吡罗昔康原料粉末、吡罗昔康固体分散体的体外溶出曲线;
图7为实施例3中卡马西平原料粉末、卡马西平和聚维酮K25和Soluplus物理混合物粉末、聚维酮K25与Soluplus物理混合物粉末、卡马西平固体分散体粉末以及卡马西平固体分散体六个月稳定性的X射线衍射图谱;
图8为实施例3中卡马西平原料粉末、卡马西平固体分散体的体外溶出曲线;
图9为实施例4中灰黄霉素原料粉末、灰黄霉素和聚维酮K25和Soluplus物理混合物粉末、聚维酮K25与Soluplus物理混合物粉末、灰黄霉素固体分散体粉末以及灰黄霉素固体分散体六个月稳定性的X射线衍射图谱;
图10为实施例4中灰黄霉素原料粉末、灰黄霉素固体分散体的体外溶出曲线;
图11为实施例5中酮康唑原料粉末、酮康唑和聚维酮K25和Soluplus物理混合物粉末、聚维酮K25与Soluplus物理混合物粉末、酮康唑固体分散体粉末以及酮康唑固体分散体六个月稳定性的X射线衍射图谱;
图12为实施例5中酮康唑原料粉末、酮康唑固体分散体的体外溶出曲线。
具体实施方式
以下通过具体实施例并结合附图对本发明的固体分散体及其制备方法和应用作进一步说明,但这并非是对本发明的限制。
以下实施例所用原料如下:
所选难溶性药物均为市售产品;
聚维酮K25:购自美国国际特品公司;
共聚维酮VA64:购自德国巴斯夫公司;
羟丙甲纤维素E5:购自美国卡乐康公司;
聚乙二醇/乙烯基己内酰胺/醋酸乙烯酯共聚物(Soluplus)、α-生育酚琥珀酸酯聚乙二醇(TPGS)、泊洛沙姆(F188):购自德国巴斯夫公司;
十二烷基硫酸钠:购自天津市福晨化学试剂厂;
胆酸钠:购自上海麦克林生物有限公司;
吐温80:购自天津市大茂化学试剂厂。
实施例1:喷雾干燥法制备二氟尼柳固体分散体
本实施例的一种二氟尼柳固体分散体,由下表所示原料通过喷雾干燥法制备得到:
其制备方法具体包括以下步骤:
(1)取二氟尼柳,表面活性性剂与水溶性高分子材料,混合均匀后加入100ml的二级水中并水浴加热至70℃,磁力搅拌直至溶液处于澄清状态,得含药的混合溶液。
(2)将所得含药的混合溶液进行喷雾干燥,喷雾干燥的进风温度为120℃,出风温度为75℃,风量为0.7m3/min,雾化压力为9kPa,液体流速为2mL/min,在喷雾干燥过程中,含药的混合溶液的温度保持在70℃。
(3)将喷雾干燥后的产物在25℃的真空干燥箱中干燥24h后过80目筛,得到二氟尼柳固体分散体粉末。
一、对本实施例制备得到的二氟尼柳固体分散体粉末进行X射线衍射测试,测试方法如下:
取适量样品置于样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
测试结果见图1,结果显示药物以无定形或分子态分散在载体材料中。
二、对本实施例制备得到的二氟尼柳固体分散体粉末进行高效液相色谱检测,检测方法如下:
精密称取相当于含有二氟尼柳约100mg的固体分散体于10mL容量瓶中,用少量甲醇溶解,轻摇至固体分散体均匀后,补加甲醇定容,作为供试品溶液。精密量取供试品溶液1mL,以甲醇稀释至10mL,从中取1mL以甲醇稀释至20mL作为对照品溶液。以水-甲醇-乙腈-冰醋酸(55:23:30:2)为流动相;检测波长为254nm。15000rpm离心15min后,取上清液5μL采用HPLC分析。记录色谱图至主峰保留时间3倍。供试品溶液如显杂质峰,以对照品溶液主峰为对照,计算杂质总含量。
测试结果显示二氟尼柳含量及有关物质在喷雾干燥前后均无明显变化,符合中国药典有关规定。
三、对本实施例制备得到的二氟尼柳固体分散体粉末进行高温高湿试验,试验方法如下:
取适量样品置于封口的离心管中,放置在40℃/75%RH的条件中,于60天取适量样品置于X射线衍射样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
试验结果见图2,结果表明以聚维酮K25为水溶性高分子载体,Soluplus为表面活性剂制备得到的二氟尼柳固体分散体的稳定性最好,编号1-4试验组均出现重结晶现象。
四、对本实施例制备得到的二氟尼柳固体分散体粉末进行体外溶出实验,方法如下:
精密称取各处方固体分散体适量(约相当于二氟尼柳10mg),满足漏槽条件,按照2010版《中国药典》附录XC桨法规定进行,转速50rpm,水浴温度37±0.5℃,溶出介质为900mL纯水。分别于5、10、20、30、45、60、90、120min取样5mL(同时补充同温等量溶出介质),经15000rpm离心15min,测定其含量。
测试结果见图3,以聚维酮K25为水溶性高分子载体,Soluplus为表面活性剂制备得到的二氟尼柳固体分散体的溶出速率较快,累计溶出度较高。其中实验编号6,也即Soluplus与聚维酮K25的质量比为2:8时,二氟尼柳固体分散体中的二氟尼柳溶出效果最好,5min溶出度达到75%,最终累计溶出度达到了98.5%。图4为实验编号6制备的二氟尼柳固体分散体与二氟尼柳原料药的溶出曲线图,结果表明本实施例的二氟尼柳固体分散体的药物累计溶出百分率显著高于二氟尼柳原料药。
实施例2:喷雾干燥法制备吡罗昔康固体分散体
本实施例的一种吡罗昔康固体分散体制备,由以下原料通过喷雾干燥法制备得到:作为活性成分的吡罗昔康、作为表面活性剂的Soluplus和作为水溶性高分子材料的聚维酮K25制备而成,其中吡罗昔康在固体分散体中的质量含量为10%,Soluplus与聚维酮K25的质量比为4:6。
其制备方法具体包括以下步骤:
(1)取300mg吡罗昔康,1.08g Soluplus与1.62g的聚维酮K25,混合均匀后加入100ml的二级水中并水浴加热至75℃,磁力搅拌直至溶液处于澄清状态,得含药的混合溶液。
(2)将所得含药的混合溶液进行喷雾干燥,喷雾干燥的进风温度为110℃,出风温度为70℃,风量为0.8m3/min,雾化压力为7kPa,液体流速为2.5mL/min,在喷雾干燥过程中,含药的混合溶液的温度保持在75℃。
(3)将喷雾干燥后的产物在25℃的真空干燥箱中干燥24h后过80目筛,得到吡罗昔康固体分散体粉末。
一、对本实施例得到的吡罗昔康固体分散体粉末进行X射线衍射,测试方法如下:
取适量样品置于样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
测试结果见图5,结果显示药物以无定形或分子态分散在载体材料中。
二、对本实施例的吡罗昔康固体分散体粉末进行高效液相色谱检测,检测方法如下:
精密称取相当于含有吡罗昔康约100mg的固体分散体于10mL容量瓶中,用少量甲醇溶解,轻摇至固体分散体均匀后,补加甲醇定容,作为供试品溶液。精密量取供试品溶液1mL,以甲醇稀释至10mL,从中取1mL以甲醇稀释至20mL作为对照品溶液。以甲醇-0.87%磷酸氢二钾缓冲溶液(60:40)为流动相;检测波长为358nm。15000rpm离心15min后,取上清液5μL采用HPLC分析。记录色谱图至主峰保留时间3倍。供试品溶液如显杂质峰,以对照品溶液主峰为对照,计算杂质总含量。
测试结果显示吡罗昔康含量及有关物质喷雾干燥前后均无明显变化,符合中国药典有关规定。
三、对本实施例的吡罗昔康固体分散体粉末进行体外溶出实验,方法如下:
精密称取各处方固体分散体适量(约相当于吡罗昔康25mg),满足漏槽条件,按照2010版《中国药典》附录XC桨法规定进行,转速100rpm,水浴温度37±0.5℃,溶出介质为900mL纯水。分别于5、10、20、30、45、60、90、120min取样5mL(同时补充同温等量溶出介质),经15000rpm离心15min,测定其含量。
测试结果见图6,结果表明本实施例的吡罗昔康固体分散体的药物累计溶出百分率显著高于吡罗昔康原料药。
实施例3:喷雾干燥法制备卡马西平固体分散体
本实施例的一种卡马西平固体分散体由以下原料通过喷雾干燥法制备得到:作为活性成分的卡马西平、作为表面活性剂的Soluplus和作为水溶性高分子材料的聚维酮K25制备而成,其中卡马西平在固体分散体中的质量含量为20%,Soluplus与聚维酮K25的质量比为2:8。
其制备方法具体包括以下步骤:
(1)取600mg卡马西平,480mg Soluplus与1.92g的聚维酮K25,混合均匀后加入100ml的二级水中并水浴加热至80℃,磁力搅拌直至溶液处于澄清状态,得含药的混合溶液。
(2)将所得含药的混合溶液进行喷雾干燥,喷雾干燥的进风温度为130℃,出风温度为85℃,风量为0.9m3/min,雾化压力为11kPa,液体流速为3mL/min,在喷雾干燥过程中,含药的混合溶液的温度保持在80℃。
(3)将喷雾干燥后的产物在25℃的真空干燥箱中干燥24h后过80目筛,得到卡马西平固体分散体粉末。
一、对本实施例制备得到的卡马西平固体分散体粉末进行X射线衍射测试,测试方法如下:
取适量样品置于样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
测试结果见图7,结果显示药物以无定形或分子态分散在载体材料中。
二、对本实施例制备得到的卡马西平固体分散体进行高效液相色谱检测,测试方法如下:
精密称取相当于含有卡马西平约100mg的固体分散体于10mL容量瓶中,用少量甲醇溶解,轻摇至固体分散体均匀后,补加甲醇定容,作为供试品溶液。精密量取供试品溶液1mL,以甲醇稀释至10mL,从中取1mL以甲醇稀释至20mL作为对照品溶液。以水-乙腈(65:35)为流动相;检测波长为284nm。15000rpm离心15min后,取上清液5μL采用HPLC分析。记录色谱图至主峰保留时间3倍。供试品溶液如显杂质峰,以对照品溶液主峰为对照,计算杂质总含量。
测试结果显示卡马西平含量及有关物质在喷雾干燥前后均无明显变化,符合中国药典有关规定。
三、对本实施例制备得到的卡马西平固体分散体进行体外溶出实验,方法如下:
精密称取各处方固体分散体适量(约相当于卡马西平15mg),满足漏槽条件,按照2010版《中国药典》附录XC桨法规定进行,转速100rpm,水浴温度37±0.5℃,溶出介质为900mL纯水。分别于5、10、20、30、45、60、90、120min取样5mL(同时补充同温等量溶出介质),经15000rpm离心15min,测定其含量。
测试结果见图8,结果表明本实施例的卡马西平固体分散体的药物累计溶出百分率显著高于卡马西平原料药。
实施例4:喷雾干燥法制备灰黄霉素固体分散体
本实施例的一种灰黄霉素固体分散体由以下原料通过喷雾干燥法制备得到,由作为活性成分的灰黄霉素、作为表面活性剂的Soluplus和作为水溶性高分子材料的聚维酮K25制备而成,其中,灰黄霉素在固体分散体的的载药质量含量为5%,Soluplus与聚维酮K25的质量比为3:17。
其制备方法具体包括以下步骤:
(1)取150mg灰黄霉素,428mg Soluplus与2.42g的聚维酮K25,混合均匀后加入100ml的二级水中并水浴加热至70℃,磁力搅拌直至溶液处于澄清状态,得含药的混合溶液。
(2)将所得含药的混合溶液进行喷雾干燥,喷雾干燥的进风温度为100℃,出风温度为65℃,风量为0.9m3/min,雾化压力为13kPa,液体流速为2mL/min,在喷雾干燥过程中,含药的混合溶液的温度保持在70℃。
(3)将喷雾干燥后的产物在25℃的真空干燥箱中干燥24h后过80目筛,得到灰黄霉素固体分散体粉末。
一、对本实施例制备得到的灰黄霉素固体分散体粉末进行X射线衍射测试,测试方法如下:
取适量样品置于样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
测试结果见图9,结果显示药物以无定形或分子态分散在载体材料中。
二、对本实施例制备得到的灰黄霉素固体分散体粉末进行高效液相色谱检测,测试方法如下:
精密称取相当于含有灰黄霉素约100mg的固体分散体于10mL容量瓶中,用少量甲醇溶解,轻摇至固体分散体均匀后,补加甲醇定容,作为供试品溶液。精密量取供试品溶液1mL,以甲醇稀释至10mL,从中取1mL以甲醇稀释至20mL作为对照品溶液。以水-乙腈(57:43)为流动相;检测波长为276nm。15000rpm离心15min后,取上清液5μL采用HPLC分析。记录色谱图至主峰保留时间3倍。供试品溶液如显杂质峰,以对照品溶液主峰为对照,计算杂质总含量。
测试结果显示灰黄霉素含量及有关物质在喷雾干燥前后均无明显变化,符合中国药典有关规定。
三、对本实施例制备得到的灰黄霉素固体分散体粉末进行体外溶出实验,方法如下:
精密称取各处方固体分散体适量(约相当于灰黄霉素10mg),满足漏槽条件,按照2010版《中国药典》附录XC桨法规定进行,转速100rpm,水浴温度37±0.5℃,溶出介质为900mL纯水。分别于5、10、20、30、45、60、90、120min取样5mL(同时补充同温等量溶出介质),经15000rpm离心15min,测定其含量。
测试结果见图10,结果表明本实施例的灰黄霉素固体分散体的药物累计溶出百分率显著高于灰黄霉素原料药。
实施例5:喷雾干燥法制备酮康唑固体分散体
本实施例的一种无酮康唑固体分散体由以下原料通过喷雾干燥法制备得到:作为活性成分的酮康唑、作为表面活性剂的Soluplus和作为水溶性高分子材料的聚维酮K25制备而成,其中,酮康唑在固体分散体中的质量含量为15%,Soluplus与聚维酮K25的质量比为1:9。
其制备方法具体包括以下步骤:
(1)取450mg酮康唑,255mg Soluplus与2.295g的聚维酮K25,混合均匀后加入100ml的二级水中并水浴加热至85℃,磁力搅拌直至溶液处于澄清状态,得含药的混合溶液。
(2)将所得含药的混合溶液进行喷雾干燥,喷雾干燥的进风温度为120℃,出风温度为75℃,风量为0.8m3/min,雾化压力为11kPa,液体流速为2.5mL/min,在喷雾干燥过程中,含药的混合溶液的温度保持在85℃。
(3)将喷雾干燥后的产物在25℃的真空干燥箱中干燥24h后过80目筛,得到酮康唑固体分散体粉末。
一、对本实施例制备得到的酮康唑固体分散体粉末进行X射线衍射测试,测试方法如下:
取适量样品置于样品架中进行测定。工作条件为采用Cu靶Kα射线;石墨单色器衍射束单色比;加速电压30kV;加速电流30mA;扫描2θ角度为3-40°;扫描速度5°/min;扫描步长0.02°。
测试结果见图11,结果显示药物以无定形或分子态分散在载体材料中。
二、对本实施例制备得到的酮康唑固体分散体粉末进行高效液相色谱检测,测试方法如下:
精密称取相当于含有酮康唑约100mg的固体分散体于10mL容量瓶中,用少量甲醇溶解,轻摇至固体分散体均匀后,补加甲醇定容,作为供试品溶液。精密量取供试品溶液1mL,以甲醇稀释至10mL,从中取1mL以甲醇稀释至20mL作为对照品溶液。以乙腈-磷酸缓冲液(62:48)为流动相;检测波长为263nm。15000rpm离心15min后,取上清液5μL采用HPLC分析。记录色谱图至主峰保留时间3倍。供试品溶液如显杂质峰,以对照品溶液主峰为对照,计算杂质总含量。
测试结果显示酮康唑含量及有关物质在喷雾干燥前后均无明显变化,符合中国药典有关规定。
三、对本实施例制备得到的酮康唑固体分散体粉末进行体外溶出实验,方法如下:
精密称取各处方固体分散体适量(约相当于酮康唑20mg),满足漏槽条件,按照2010版《中国药典》附录XC桨法规定进行,转速100rpm,水浴温度37±0.5℃,溶出介质为900mL纯水。分别于5、10、20、30、45、60、90、120min取样5mL(同时补充同温等量溶出介质),经15000rpm离心15min,测定其含量。
测试结果见图12,结果表明本实施例的酮康唑固体分散体的药物累计溶出百分率显著高于酮康唑原料药。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!