制备蛋白质‑聚氨基酸偶联物的方法与流程
本公开涉及生物医药领域,具体地涉及制备位点特异性蛋白质-聚氨基酸偶联物的方法。
背景技术:
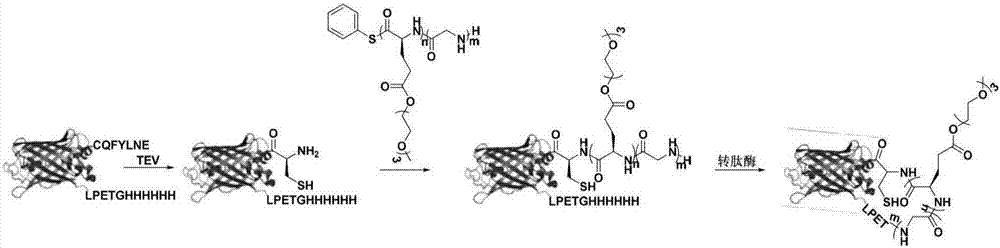
:蛋白质药物(如抗体、干扰素)已经广泛用于生物医药领域,如靶向治疗、临床诊断等。相比于其他药物,蛋白质药物特异性较好,见效快,毒性低。然而,其稳定性差、血液循环时间短等缺点在一定程度上限制了此类药物的药效和应用。目前,研究较多的提高蛋白质稳定性,延长其血液循环时间的体系是蛋白质-高分子杂化体系。蛋白质药物往往由于体内蛋白酶的降解致使其血液循环时间比较短暂,因此将水溶性较好的高分子材料接枝在蛋白质表面,对蛋白质有一定的保护作用,既可降低蛋白质的免疫原性,也可以降低蛋白酶对蛋白质类药物的降解以延长其血液循环时间。举例而言,干扰素-α2a是治疗慢性丙型肝炎的药物,在其表面接枝一条40kda的聚乙二醇链将会显著延长其血液循环时间,干扰素-α2a-聚乙二醇杂化材料已应用于临床。然而,目前临床中已应用的蛋白质-高分子杂化体系中主要使用聚乙二醇作为高分子片段。聚乙二醇具有免疫原性低、水溶性好等优点,但随着聚乙二醇在化妆品、食品、制药等领域的广泛应用,大部分人的体内已产生聚乙二醇抗体,反而会加速聚乙二醇修饰的蛋白质类药物在血液中的清除。更严重的是聚乙二醇在人体内不可降解,长期服用则会导致积累毒性。因此,寻找可降解、生物相容性好的高分子材料迫在眉睫。聚氨基酸是一类具有良好生物相容性的高分子材料,其主链的可降解性及侧链的修饰便利性都使得它在蛋白质改性方面具有广泛的应用。更重要的是聚氨基酸具有多肽主链结构、生物相容性好、免疫原性低等特点,非常适合用于蛋白质药物的修饰。由此,本公开了提供了一种蛋白质-聚氨基酸偶联物及一种蛋白质-聚氨基酸环状偶联物以克服本领域尚存在的不足之处。技术实现要素:在本公开的一方面,提供了制备位点特异性蛋白质-聚氨基酸偶联物的方法,包括:(1)利用引发剂引发n-羧基内酸酐聚合,得到具有碳结构的聚氨基酸;(2)将所得到的聚氨基酸与含有1,2-巯基乙胺结构的蛋白质混合,发生自然化学连接反应,得到位点特异性蛋白质-聚氨基酸偶联物。根据本公开的一实施方案,在步骤(1)中,利用引发剂r5xy引发n-羧基内酸酐聚合,得到含有碳结构的聚氨基酸paa-xr5,如下所示:其中:x表示硫或硒;y代表氢或三烷基硅基,其中三烷基硅基中的烷基为c1-c10烷基;r1每次出现时独立地表示天然氨基酸侧链或非天然氨基酸侧链;r5表示取代或未取代的c1-c30烷基、取代或未取代的c2-c30烯基、取代或未取代的c2-c30炔基、取代或未取代的c3-c30环烷基、取代或未取代的c3-c30环烯基、取代或未取代的c1-c30杂烷基、取代或未取代的c2-c30杂烯基、取代或未取代的c2-c30杂炔基、取代或未取代的c1-c30杂环烷基、取代或未取代的c2-c30杂环烯基、取代或未取代的c6-c30芳基、取代或未取代的c5-c30杂芳基;以及n为选自1至200的整数。根据本公开的另一实施方案,在步骤(1)中,利用引发剂r5xy引发n-羧基内酸酐聚合,得到含有碳结构的聚氨基酸paa-xr5,如下所示:其中:x表示硫或硒;y代表氢或三烷基硅基,其中三烷基硅基中的烷基为c1-c10烷基;r1每次出现时独立地表示天然氨基酸侧链或非天然氨基酸侧链;r2每次出现时独立地表示氢或c1-c10烷基、c2-c10烯基、c2-c10炔基;r5表示取代或未取代的c1-c30烷基、取代或未取代的c2-c30烯基、取代或未取代的c2-c30炔基、取代或未取代的c3-c30环烷基、取代或未取代的c3-c30环烯基、取代或未取代的c1-c30杂烷基、取代或未取代的c2-c30杂烯基、取代或未取代的c2-c30杂炔基、取代或未取代的c1-c30杂环烷基、取代或未取代的c2-c30杂环烯基、取代或未取代的c6-c30芳基、取代或未取代的c5-c30杂芳基;以及n为选自1至200的整数。根据本公开的又一实施方案,r5xy为三甲基硅基苯硫酚或三甲基硅基苯硒酚。根据本公开的再一实施方案,步骤(1)在非质子性溶剂中且在室温下进行。根据本公开的一实施方案,对步骤(1)中所得的聚氨基酸进行后修饰,所述后修饰包括将所得的聚氨基酸与调节剂混合并在紫外灯照射下反应,所述调节剂为巯基乙胺盐酸盐或巯基丙酸。根据本公开的一实施方案,在步骤(2)中,将步骤(1)中得到的聚氨基酸与含有1,2-巯基乙胺结构的蛋白质混合,室温下发生自然化学连接反应,得到位点特异性蛋白质-聚氨基酸偶联物,如下所示:其中:ptn表示蛋白质。根据本公开的另一实施方案,在步骤(2)中,将步骤(1)中得到的聚氨基酸与含有1,2-巯基乙胺结构的蛋白质混合,室温下发生自然化学连接反应,得到位点特异性蛋白质-聚氨基酸偶联物,如下所示:其中:ptn表示蛋白质。在本公开的又一方面,提供了由上述方法制备的蛋白质-聚氨基酸偶联物。在本公开的另一方面,提供了由上述方法制备的蛋白质-聚氨基酸偶联物在制造蛋白质类药物中的用途。附图说明在下文参考附图来进一步描述本文所例示的实施方案,但是所述附图仅仅是为了让本领域技术人员更好地理解本公开内容,而不旨在限定该公开内容的范围。图1为根据本公开一实施方案的位点特异性蛋白质-聚氨基酸偶联物示意图;图2为根据本公开一实施方案的蛋白质-聚氨基酸环化反应示意图;图3为根据实施例1的聚氨基酸p1的maldi-tof谱图;图4为根据实施例15的环状偶联物maldi-tof谱图,其中o表示l-谷氨酸(乙二醇)3酯链段,g表示甘氨酸链段,角标表示相应链段的聚合度;图5为根据实施例16的enlyfq-cys-egfp蛋白的uplc-ms谱图;图6为根据实施例18的cys-egfp蛋白的uplc-ms谱图;图7为根据实施例19的聚氨基酸p1(n=7)-增强型绿色荧光蛋白偶联物的电泳图,其示出变性胶蛋白质凝胶电泳(a)及非变性胶蛋白质凝胶电泳(b);图8为根据实施例20的enlyfqc-增强型绿色荧光蛋白-聚乙二醇-聚甘氨酸偶联物的电泳图,其示出变性胶蛋白质凝胶电泳(a)及非变性胶蛋白质凝胶电泳(b);图9为根据实施例21的聚氨基酸p1(n=7)-增强型绿色荧光蛋白-聚甘氨酸-聚乙二醇节点状偶联物的电泳图,其示出变性胶蛋白质凝胶电泳(a)及非变性胶蛋白质凝胶电泳(b);图10为根据实施例22的增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)-干扰素α哑铃状偶联物的电泳图,其示出变性胶蛋白质凝胶电泳(a)及非变性胶蛋白质凝胶电泳(b);图11为根据实施例23的环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物的表征:maldi-tof(a),变性胶蛋白质凝胶电泳(b)及免疫印迹分析(c);图12为根据实施例24的cys-增强型绿色荧光蛋白(a)、聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白(b)和环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物(c)酶稳定性的表征:图a、b和c内的方框为蛋白质或蛋白质-聚氨基酸偶联物原料;图13为根据实施例26的蛋白-干扰素的uplc-ms谱图;图14为根据实施例27和28的聚氨基酸偶联物的电泳图;图15为根据实施例29的环状(聚氨基酸p2(n=7或20,m=3)-干扰素α-lpetgleh6)偶联物的表征:a.环状(聚氨基酸p2(n=7,m=3)-干扰素α-lpetgleh6)偶联物的变性胶凝胶电泳;b.环状(聚氨基酸p2(n=7,m=3)-干扰素α-lpetgleh6)偶联物的免疫印迹分析;c.环状(聚氨基酸p2(n=20,m=3)-干扰素α-lpetgleh6)偶联物的变性胶凝胶电泳;d.环状(聚氨基酸p2(n=20,m=3)-干扰素α-lpetgleh6)偶联物的免疫印迹分析;图16为示出根据实施例30的各蛋白质样品的tm值的柱形图;图17为示出根据实施例31的胰蛋白酶耐受性测试的结果的图表;图18为根据实施例32的wt-ifnα,聚氨基酸p1(n=100,l型)-ifnα偶联物与聚氨基酸p1(n=100,d,l型)-ifnα偶联物的圆二色谱表征;图19为示出根据实施例34的聚氨基酸-干扰素偶联物的血药浓度的图表;图20a和b为示出根据实施例35的聚氨基酸-偶联物与环状聚氨基酸-偶联物的体内抗肿瘤活性的图表;图21为根据实施例37的聚氨基酸-抗体偶联物的电泳图。具体实施方式在下文中,将根据具体实施方案来进一步阐述本公开的内容。然而,所列举的具体实施方案仅出于例示目的,而并不旨在限制本公开的内容。本领域技术人员会认识到,以下任一实施方案中的具体特征可以用于任何其他实施方案,只要其不背离本公开的主旨即可。定义在下文中,除非另外定义,本文使用的所有术语(包括技术术语和科学术语)均具有如本公开所属领域的普通技术人员通常理解的相同含义。术语,诸如在常用词典中定义的那些术语,应解释为具有与其在相关技术的上下文中的含义相符的含义,并且不会以理想化或过于正式的含义来解释,除非本文明确如此定义。如本文所用,术语“c1-30”是指基团的主链中具有1至30的范围内的任意整数值的碳原子,例如1、2、3、4、5、6、7、8、9、10、12、15、18、20、30个碳原子。如本文所用,术语“烷基”是指具有直链或支链的饱和脂肪族烃基。其非限制性实例包括甲基、乙基、丙基、正丁基、叔丁基、戊基、己基、庚基、辛基、壬基、癸基等。如本文所用,术语“烯基”是指在沿着烷基的碳链的一个或多个位置处具有至少一个碳-碳双键的烃基。其非限制性实例包括乙烯基、丙烯基、丁烯基等。如本文所用,术语“炔基”是指在沿着烷基的碳链的一个或多个位置处具有至少一个碳-碳叁键的烃基。其非限制性实例包括乙炔基、丙炔基等。如本文所用,术语“杂烷基”、“杂烯基”、“杂炔基”分别是指包含至少一个选自n、o、si、p和s的杂原子的烷基、烯基、炔基。如本文所用,术语“环烷基”是指单环饱和烃基。其非限制性实例包括环丙基、环丁基、环戊基、环己基和环庚基。如本文所用,术语“杂环烷基”是指包含作为成环原子的至少一个选自n、o、si、p和s的杂原子的单碳环基团。其非限制性实例包括四氢呋喃基和四氢噻吩基。如本文所用,术语“环烯基”是指在其环中具有碳原子和至少一个双键的单环基团,而且其不是芳香族的。其非限制性实例包括环戊烯基、环己烯基和环庚烯基。如本文所用,术语“杂环烯基”是指在其环中包括作为成环原子的至少一个选自n、o、si、p和s的杂原子和至少一个双键的单碳环基团。其非限制性实例包括4,5-二氢-1,2,3,4-噁三唑基、2,3-二氢呋喃基和2,3-二氢噻吩基。如本文所用,术语“芳基”是指包含碳环芳香族体系的基团。其非限制性实例包括苯基、萘基、蒽基、菲基、芘基等。当芳基包括多个环时,各自的环可以彼此稠合。如本文所用,术语“杂芳基”是指具有包含作为成环原子的至少一个选自n、o、si、p和s的杂原子的碳环芳香族体系的基团。其非限制性实例包括吡啶基、嘧啶基、吡嗪基、哒嗪基、三嗪基、喹啉基、异喹啉基等。当杂芳基包括多个环时,各自的环可以彼此稠合。蛋白质-聚氨基酸偶联物在本公开的一实施方案,提供了蛋白质-聚氨基酸偶联物,其具有如通式1表示的结构:其中:ptn表示蛋白质;et当与ptn的n端连接时,为而当与ptn的除n端之外的位点连接时,为r1每次出现时独立地表示天然氨基酸侧链或非天然氨基酸侧链;r2每次出现时独立地表示氢或c1-c10烷基、c2-c10烯基、c2-c10炔基;n为选自1至300的整数。根据本公开的一实施方案,ptn是选自酶、激素、细胞因子、单克隆抗体和/或蛋白质疫苗的蛋白质。根据本公开的另一实施方案,ptn是蛋白质类药物,如多肽激素、单克隆抗体、干扰素、白介素、集落刺激因子、重组疫苗等。根据本公开的另一实施方案,ptn是干扰素α2b。可适用于本文的蛋白类药物不受特别的限制,只要其能够与et连接基团连接即可。理论上,任何蛋白质类药物经修饰后均可适用于本文。根据本公开的一实施方案,可用于本公开的酶包括但不限于:蛋白水解酶、淀粉酶、脂肪酶、纤维素酶、胰蛋白酶、胰凝乳蛋白酶、链激酶、尿激酶、纤溶酶、凝血酶、谷氨酰胺酶、精氨酸酶、丝氨酸脱水酶、苯丙氨酸氨解酶、亮氨酸脱氢酶、青霉素酶、超氧化物歧化酶、葡聚糖酶和/或右旋糖酐酶。根据本公开的一实施方案,可用于本公开的激素包括但不限于下丘脑激素、垂体激素、胃肠激素、胰岛素、降钙素。根据本公开的一实施方案,可用于本公开的细胞因子包括但不限于白细胞介素、干扰素、集落刺激因子、趋化因子和/或生长因子。根据本公开的一实施方案,可用于本公开的白细胞介素包括il-1、il-2、il-3、il-4、il-5、il-6、il-7、il-8、il-9、il-10、il-11、il-12、il-13、il-14、il-15、il-16、il-17、il-18、il-19、il-20、il-21、il-22、il-23、il-24、il-25、il-26、il-27、il-28、il-29、il-30、il-31和/或il-32。根据本公开的一实施方案,可用于本公开的干扰素包括但不限于ifn-α、ifn-β、ifn-γα、ifn-λ及其亚型。根据本公开的一实施方案,可用于本公开的集落刺激因子包括但不限于:粒细胞集落刺激因子、巨噬细胞集落刺激因子、粒细胞-巨噬细胞集落刺激因子、多能集落刺激因子、干细胞因子、白血病抑制因子和/或红细胞生成素。根据本公开的一实施方案,可用于本公开的生长因子包括但不限于表皮细胞生长因子、转化生长因子、胰岛素样生长因子和/或神经生长因子。根据本公开的一实施方案,可用于本公开的单克隆抗体包括但不限于:曲妥珠单抗、西妥昔单抗、达利珠单抗、他尼珠单抗、阿巴伏单抗、阿德木单抗、阿夫土珠单抗、阿仑单抗、培化阿珠单抗、阿麦妥昔单抗、阿泊珠单抗、巴维昔单抗、贝妥莫单抗、贝利木单抗、贝伐单抗、莫-比伐珠单抗、贝伦妥单抗-维多汀、莫-坎妥珠单抗、拉-坎妥珠单抗、卡罗单抗-喷地肽、卡妥索单抗、泊-西他珠单抗、西妥木单抗、可那木单抗、达西珠单抗、达洛珠单抗、地莫单抗、依美昔单抗、依决洛单抗、依洛珠单抗、恩司昔单抗、依帕珠单抗、厄马索单抗、埃达珠单抗、法勒珠单抗、芬妥木单抗、加利昔单抗、吉妥珠单抗、吉妥珠单抗、吉瑞昔单抗、格莱木单抗-维多汀、替-伊莫单抗、伊戈伏单抗、拉-英达西单抗、英妥木单抗、伊珠单抗-奥佐米星、伊匹木单抗、伊妥木单抗、拉贝珠单抗、来沙木单抗、林妥珠单抗、莫-洛伏珠单抗、鲁卡木单抗、鲁昔单抗、马帕木单抗、马妥珠单抗、米拉珠单抗、米妥莫单抗、莫加珠单抗、他那可单抗、他那莫单抗、奈昔木单抗、尼妥珠单抗、纳武单抗、奥法木单抗、奥纳珠单抗、莫-奥珠单抗、奥戈伏单抗、帕尼单抗、帕曲土单抗、培妥珠单抗、普立木单抗、雷妥莫单抗、雷莫芦单抗、利妥木单抗、利妥昔单抗、罗妥木单抗、奥马珠单抗、西罗珠单抗、司妥昔单抗、帕-他莫单抗、替妥莫单抗、替妥木单抗、替西木单抗、曲美木单抗、替加珠单抗、托西莫单抗、西莫白介素单抗、乌瑞鲁单抗、维妥珠单抗、伏洛昔单抗、伏妥莫单抗和扎芦木单抗,包括其抗原结合片段。根据本公开的一实施方案,可用于本公开的蛋白质疫苗包括但不限于白喉类毒素、破伤风类毒素、炭疽分泌蛋白疫苗和/或乙型肝炎血源疫苗。根据本公开的一实施方案,r1每次出现时表示天然氨基酸侧链,所述天然氨基酸选自甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、酪氨酸、色氨酸、丝氨酸、苏氨酸、半胱氨酸、蛋氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸或组氨酸。根据本公开的另一实施方案,r1每次出现时表示非天然氨基酸侧链,如经人工修饰的上述天然氨基酸。根据本公开的又一实施方案,r1每次出现时表示经寡聚乙二醇(聚合度为2-10,如二缩三乙二醇,oeg3)修饰、磷酸酯修饰、丙烯氧基苄酯修饰、烯丙基二缩三乙二醇修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸的侧链。根据本公开的再一实施方案,r1每次出现时表示经二缩三乙二醇(oeg3)修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸的侧链。本文对r1没有特别的限制,理论上,任何天然氨基酸可以直接地用于本公开的偶联物,或者经修饰后,再用于本公开的偶联物,以便改善稳定性。根据本公开的一实施方案,r2每次出现时表示氢或c1-c10烷基。根据本公开的另一实施方案,r2每次出现时表示氢、甲基、乙基、正丙基、异丙基、正丁基、叔丁基、戊基、己基。根据本公开的又一实施方案,r2表示氢或甲基。根据本公开的一实施方案,n表示选自1至200的整数。根据本公开的另一实施方案,n表示选自1至150的整数。根据本公开的又一实施方案,n表示选自1至100的整数。根据本公开的再一实施方案,n表示选自1至50的整数。在理论上,n可以为任何数值,只要所得的偶联物稳定存在即可,本领域技术人员可以根据实际应用,设定具体的n值,而且,这种选择应在本领域技术人员的能力范围内。根据本公开的一实施方案,提供了蛋白质-聚氨基酸偶联物,其具有如通式2表示的结构:其中:ptn、et、r1和r2如上文对通式1所定义;n为选自1至200的整数;以及m为选自1至30的整数。根据本公开的一实施方案,n表示选自1至150的整数。根据本公开的另一实施方案,n表示选自1至100的整数。根据本公开的又一实施方案,n表示选自1至50的整数。根据本公开的一实施方案,m表示选自1至20的整数。根据本公开的另一实施方案,m表示选自1至10的整数。如上所述,n值和m值可以为任何数值,并不局限于本文所列举的特定范围,条件是所得的偶联物可以稳定存在。本领域技术人员可以根据实际应用,设定具体的n值和m值,而且,这种选择应在本领域技术人员的能力范围内。蛋白质-聚氨基酸环状偶联物根据本公开的一实施方案,提供了蛋白质-聚氨基酸环状偶联物,其具有如通式3表示的结构:其中:ptn表示蛋白质,其具有n端处的半胱氨酸残基和c端处的lpxat序列,其中xa表示一种或多种氨基酸;paa(gly)m为具有通式4表示的结构:其中:r1每次出现时独立地表示天然氨基酸侧链或非天然氨基酸侧链;r2每次出现时独立地表示氢或c1-c10烷基、c2-c10烯基、c2-c10炔基;n为选自10至300的整数;以及m为选自1至30的整数。根据本公开的一实施方案,ptn是上文所述蛋白质类药物,其中所述蛋白质类药物具有n端处的半胱氨酸残基和c端处的lpxat序列,或可以经修饰而具有n端处的半胱氨酸残基和c端处的lpxat序列。根据本公开的另一实施方案,ptn是n端处具有半胱氨酸残基且c端处具有lpxat序列的干扰素α2b。根据本公开的又一实施方案,ptn是具有氨基酸序列seqidno.1的蛋白质。根据本公开的再一实施方案,ptn如上文关于通式1所述。根据本公开的一实施方案,r1每次出现时表示天然氨基酸侧链,所述天然氨基酸选自甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、酪氨酸、色氨酸、丝氨酸、苏氨酸、半胱氨酸、蛋氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸或组氨酸。根据本公开的另一实施方案,r1每次出现时表示非天然氨基酸侧链,如经人工修饰的上述天然氨基酸侧链。根据本公开的又一实施方案,r1每次出现时表示经寡聚乙二醇(聚合度为2-10,如二缩三乙二醇,oeg3)修饰、磷酸酯修饰、丙烯氧基苄酯修饰、烯丙基二缩三乙二醇修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸侧链。根据本公开的再一实施方案,r1每次出现时表示经二缩三乙二醇(oeg3)修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸侧链。根据本公开的一实施方案,xa可以是选自甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、酪氨酸、色氨酸、丝氨酸、苏氨酸、半胱氨酸、蛋氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸或组氨酸中的一种或多种。根据本公开的一实施方案,r2每次出现时表示氢或c1-c10烷基。根据本公开的另一实施方案,r2每次出现时表示氢、甲基、乙基、正丙基、异丙基、正丁基、叔丁基、戊基、新戊基、己基。根据本公开的又一实施方案,r2表示氢。根据本公开的一实施方案,n表示选自10至200的整数。根据本公开的另一实施方案,n表示选自10至150的整数。根据本公开的又一实施方案,n表示选自10至100的整数。根据本公开的再一实施方案,n表示选自10至50的整数。根据本公开的一实施方案,m表示选自1至20的整数。根据本公开的另一实施方案,m表示选自1至10的整数。如上所述,n值和m值可以为任何数值,并不局限于本文所列举的特定范围,条件是所得的偶联物可以稳定存在。本领域技术人员可以根据实际应用,设定具体的n值和m值,而且,这种选择应在本领域技术人员的能力范围内。用于蛋白(环化)偶联的聚氨基酸根据本公开的一实施方案,提供了用于蛋白(环化)偶联的聚氨基酸,其具有如通式5所示的结构:其中:x表示硫或硒;r1每次出现时独立地表示天然氨基酸侧链或非天然氨基酸侧链;r2每次出现时独立地表示氢或c1-c10烷基、c2-c10烯基、c2-c10炔基;r5表示取代或未取代的c1-c30烷基、取代或未取代的c2-c30烯基、取代或未取代的c2-c30炔基、取代或未取代的c3-c30环烷基、取代或未取代的c3-c30环烯基、取代或未取代的c1-c30杂烷基、取代或未取代的c2-c30杂烯基、取代或未取代的c2-c30杂炔基、取代或未取代的c1-c30杂环烷基、取代或未取代的c2-c30杂环烯基、取代或未取代的c6-c30芳基、取代或未取代的c5-c30杂芳基;以及n为选自1至200的整数。根据本公开的一实施方案,r1每次出现时表示天然氨基酸侧链,所述天然氨基酸选自甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、酪氨酸、色氨酸、丝氨酸、苏氨酸、半胱氨酸、蛋氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸或组氨酸。根据本公开的另一实施方案,r1每次出现时表示非天然氨基酸侧链,如经人工修饰的上述天然氨基酸侧链。根据本公开的又一实施方案,r1每次出现时表示经寡聚乙二醇(聚合度为2-10,如二缩三乙二醇,oeg3)修饰、磷酸酯修饰、丙烯氧基苄酯修饰、烯丙基二缩三乙二醇修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸侧链。根据本公开的再一实施方案,r1每次出现时表示经二缩三乙二醇(oeg3)修饰的酪氨酸、丝氨酸、苏氨酸、半胱氨酸、天冬氨酸和/或谷氨酸侧链。根据本公开的一实施方案,r2每次出现时表示氢或c1-c10烷基。根据本公开的另一实施方案,r2每次出现时表示氢、甲基、乙基、正丙基、异丙基、正丁基、叔丁基、戊基、新戊基、己基。根据本公开的又一实施方案,r2表示氢。根据本公开的一实施方案,r5表示取代或未取代的c1-c10烷基、取代或未取代的c2-c10烯基、取代或未取代的c2-c10炔基、取代或未取代的c3-c10环烷基、取代或未取代的c3-c10环烯基、取代或未取代的c1-c10杂烷基、取代或未取代的c2-c10杂烯基、取代或未取代的c1-c10杂环烷基、取代或未取代的c2-c10杂环烯基、取代或未取代的c6-c18芳基、取代或未取代的c5-c18杂芳基。根据本公开的另一实施方案,r5表示取代或未取代的c1-c10烷基、取代或未取代的c3-c10环烷基、取代或未取代的c1-c10杂烷基、取代或未取代的c1-c10杂环烷基、取代或未取代的c6-c18芳基、取代或未取代的c5-c18杂芳基。根据本公开的一实施方案,用于取代r5的取代基选自卤素、卤代c1-3烷基、羟基、氨基、巯基、羰基、羧基、磺酸基、羧酸盐、磺酸盐、酯基、酰胺和/或卤素。根据本公开的另一实施方案,r5选自以下结构式,但不限于此:根据本公开的又一实施方案,r5表示甲基、乙基、丙基、正丁基、叔丁基、戊基、己基、环丙基、环丁基、环戊基、环己基、苯基。根据本公开的再一实施方案,r5表示苯基。根据本公开的一实施方案,n表示选自1至200的整数。根据本公开的另一实施方案,n表示选自1至150的整数。根据本公开的又一实施方案,n表示选自1至100的整数。根据本公开的再一实施方案,n表示选自1至50的整数。在理论上,n可以为任何数值,只要所得的聚氨基酸稳定存在即可,本领域技术人员可以根据实际应用,设定具体的n值,而且,这种选择应在本领域技术人员的能力范围内。根据本公开的一实施方案,提供了用于蛋白(环化)偶联的聚氨基酸,其具有如通式6所示的结构:其中:x、r1、r2和r5如上文对通式5所定义;n为选自10至200的整数;m为选自1至30的整数。根据本公开的一实施方案,n表示选自10至150的整数。根据本公开的另一实施方案,n表示选自10至120的整数。根据本公开的又一实施方案,n表示选自10至100的整数。根据本公开的再一实施方案,n表示选自10至80的整数。根据本公开的其他实施方案,n表示选自10至50的整数。根据本公开的一实施方案,m表示选自1至20的整数。根据本公开的另一实施方案,m表示选自1至10的整数。如上所述,n值和m值可以为任何数值,并不局限于本文所列举的特定范围,条件是所得的偶联物可以稳定存在。本领域技术人员可以根据实际应用,设定具体的n值和m值,而且,这种选择应在本领域技术人员的能力范围内。制备蛋白质-聚氨基酸偶联物的方法根据本公开的一实施方案,提供了制备本文所述的位点特异性蛋白质-聚氨基酸偶联物的方法,包括:(1)利用引发剂引发n-羧基内酸酐聚合,得到具有碳结构的聚氨基酸;(2)将所得到的聚氨基酸与含有1,2-巯基乙胺结构的蛋白质混合,发生自然化学连接反应,得到位点特异性蛋白质-聚氨基酸偶联物。根据本公开的一实施方案,步骤(1)包括:利用引发剂(r5xy)引发n-羧基内酸酐聚合,得到含有碳结构的聚氨基酸(paa-xr5),如下所示:其中:x、r1、r5和n如上文对通式5所定义;y代表氢或三烷基硅基,其中三烷基硅基中的烷基优选为c1-c10烷基,如甲基、乙基、丙基、丁基、戊基、己基等。根据本公开的另一实施方案,当y为三烷基硅基时,所得的聚氨基酸的氮端处的三烷基硅基氨基甲酸酯结构在遇到空气中的水分时很容易脱去,因此最终得到的聚氨基酸氮端为氨基。根据本公开的又一实施方案,步骤(1)包括:利用引发剂(r5xy)引发n-羧基内酸酐聚合,得到含有碳结构的聚氨基酸(paa-xr5),如下所示:其中:x、r1、r2、r5和n如上文对通式5所定义;y代表氢或三烷基硅基,其中三烷基硅基中的烷基优选为c1-c10烷基,如甲基、乙基、丙基、丁基、戊基、己基等。根据本公开的一实施方案,以r5xy为三甲基硅基苯硫酚或三甲基硅基苯硒酚为例,反应式为:其中x、r1和n如上文对通式5所定义。根据本公开的一实施方案,在步骤(1)中,于非质子性溶剂,例如二甲基甲酰胺(dmf)、四氢呋喃(thf)、二氯甲烷(dcm)等溶剂中进行聚合。根据本公开的另一实施方案,反应温度通常为室温(25℃),反应时间则视单体和所意欲的聚合度而定,从几十分钟到几十小时不等,如20分钟、40分钟、1小时、10小时、15小时、20小时、25小时、30小时等。根据本公开的一实施方案,可以将由步骤(1)获得的含有碳结构的聚氨基酸进行后修饰,包括:将所得的聚氨基酸与调节剂混合并反应,由此得到经后修饰的聚氨基酸。根据本公开的另一实施方案,所述调节剂为巯基乙胺盐酸盐或巯基丙酸。根据本公开的又一实施方案,所述后修饰过程可在紫外灯照射下反应一段时间,例如1小时、3小时、5小时、10小时等,本领域技术人员可以根据具体反应进程而定。根据本公开的其他实施方案,后修饰在溶液中进行,例如含非质子性溶剂的溶液,如二甲基甲酰胺(dmf)、四氢呋喃(thf)、二氯甲烷(dcm)等,而且,所述溶液还可以包含光敏剂,以促进反应进行,例如安息香二甲醚(dmpa)等。根据本公开的一实施方案,步骤(2)包括:将步骤(1)中得到的聚氨基酸与含有1,2-巯基乙胺结构(如n端半胱氨酸,或者在c端或其他任意位置插入含有1,2-巯基乙胺结构的非天然氨基酸)的蛋白质混合,室温下发生自然化学连接(nativechemicalligation)反应,得到位点特异性蛋白质-聚氨基酸偶联物,如下所示:其中ptn、r1、r2和n如上文对通式1所定义。根据本公开的另一实施方案,步骤(2)包括:将步骤(1)中得到的聚氨基酸与含有1,2-巯基乙胺结构(如n端半胱氨酸,或者在c端或其他任意位置插入含有1,2-巯基乙胺结构的非天然氨基酸)的蛋白质混合,室温下发生自然化学连接反应,得到位点特异性蛋白质-聚氨基酸偶联物,如下所示:其中ptn、r1和n如上文对通式1所定义。根据本公开的一实施方案,在步骤(2)中采用ph为6.5-7.0反应溶液,通常会加入还原剂tcep(三(2-羧乙基)膦),但也不是必要的。上述方法利用r5xy引发一种n-羧基内酸酐聚合,得到含有碳结构的聚氨基酸paa-xr5,paa-xr5再与含有1,2-巯基乙胺结构的蛋白质混合,室温下发生自然化学连接反应,得到位点特异的蛋白质-聚氨基酸偶联物。以三甲基硅基苯硫酚引发n-羧基内酸酐聚合为例,该过程如图1所示。通过标准的分子克隆方法可以在蛋白质的n端插入半胱氨酸-甘氨酸(n末端为半胱氨酸残基,甘氨酸残基作为连接增加n端的灵活性),或者利用tag和琥珀酰氨酰合成酶/trna的方法(参见文献nguyen,d.p.;elliott,t.;holt,m.;muir,t.w.;chin,j.w.,geneticallyencoded1,2-aminothiolsfacilitaterapidandsite-specificproteinlabelingviaabio-orthogonalcyanobenzothiazolecondensation.jamchemsoc2011,133(30),11418-21.)插入侧链带有1,2-巯基乙胺结构的非天然氨基酸,以进行后续的位点特异的蛋白质-聚氨基酸偶联反应。制备蛋白质-聚氨基酸环状偶联物的方法根据本公开的一实施方案,提供了制备本文所述的位点特异性蛋白质-聚氨基酸环状偶联物的方法,包括:(1)利用引发剂引发n-羧基内酸酐和甘氨酸的n-羧基内酸酐聚合,得到c端具有苯基硫酯结构、n端具有聚甘氨酸嵌段结构的嵌段聚氨基酸;(2)将所得到的嵌段聚氨基酸与n端半胱氨酸、c端具有lpxatg序列的蛋白质混合,连续发生自然化学连接反应和转肽酶反应,实现蛋白质的环化。根据本公开的一实施方案,步骤(1)包括:利用引发剂(r5xy)引发n-羧基内酸酐和甘氨酸的n-羧基内酸酐聚合,得到c端具有苯基硫酯结构、n端具有聚甘氨酸嵌段结构的嵌段聚氨基酸,如下所示:其中r1、r5、x、y、m和n如上文对通式6所定义。根据本公开的另一实施方案,步骤(1)包括:利用引发剂(r5xy)引发n-羧基内酸酐和另一种n-羧基内酸酐聚合,得到c端具有苯基硫酯结构、n端具有聚甘氨酸嵌段结构的嵌段聚氨基酸,如下所示:其中x、r1、r2、r5、n和m如上文对通式6所定义;y代表氢或三烷基硅基,其中三烷基硅基中的烷基优选为c1-c10烷基,如甲基、乙基、丙基、丁基、戊基、己基等。根据本公开的一实施方案,步骤(2)包括:将步骤(1)得到的嵌段聚氨基酸(以h-(gly)mpaa-xr5为例)与n端半胱氨酸、c端具有lpxatg(xa为任一氨基酸)序列的蛋白质混合,连续发生自然化学连接反应和转肽酶反应,实现蛋白质的环化,如下所示:上述lpxatg序列是转肽酶的识别位点,嵌段聚氨基酸与所述蛋白质的氮端半胱氨酸发生连接反应,然后在转肽酶的作用下实现环化。为便于蛋白质的后续纯化,可以在反应前的蛋白质c末端加上多聚组氨酸标签(his-tag),环化后通过镍亲和层析的方法纯化。根据本公开的一个实施方案,通过质粒构建在egfp基因序列的n端插入烟草蚀纹病毒蛋白酶(tobaccoetchvirusprotease,tev)的酶切序列enlyfq,在其c端插入lpetg序列,以进行tev催化的偶联反应,具体的蛋白质氨基酸序列见序列表中seqidno:1。嵌段聚氨基酸c端通过自然化学连接与n端带有半胱氨酸的蛋白质进行偶联,其n端在转肽酶的作用下与蛋白质c端的lpetg偶联,其反应如图2所示。根据本公开聚合而得的氨基酸序列可以由任意水溶性氨基酸组成,涵盖天然、非天然、l型、d型氨基酸等,因此较生物表达的方式而言具有显著的材料多样性优势。同时,该聚合得到的α-聚氨基酸具有可控的分子量及分子量分布,因此可以根据实际需求来制备不同长度的多肽材料。根据本公开的方法可以原位赋予α-聚氨基酸c端的硫酯和n端的重复甘氨酸序列,它们可以通过非常高效的反应与蛋白质相连,由此使得蛋白质修饰成本大大降低,有着广阔的应用前景。根据本公开的蛋白质-高分子杂化材料不仅继承了传统的蛋白质-高分子杂化材料的优点,如将具有特殊性质(如热响应性、光响应性等)的高分子于蛋白质的连接,使得蛋白质-高分子杂化材料具有一定的功能性和响应性。此外,蛋白质的降解通常是在c端或者n端发生,通过高分链将其两端连接进行环化可以降低蛋白质被降解的速率,增加其稳定性,延长其血液循环时间。因此,本公开为蛋白质药物提供了十分广阔的应用前景。实施例以下的实施例便于更好的理解本公开的内容,而不旨在对其作出任何限定。下述实施例中所使用的实验方法如无特殊说明,均为常规方法,所用的材料、试剂等,如无特殊说明,均可从商业途径得到,所利用的质粒均通过标准分子克隆的方法得到。实施例1:用于蛋白偶联的聚氨基酸p1的合成在手套箱中将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐(n1,40.0mg,0.131mmol,20当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(13.6μl×0.5m,1.0当量),室温搅拌25小时后将反应液倒入40ml乙醚溶液中,析出的白色沉淀即为产品。离心得到固体产品,倾倒出上清液并再次用40ml乙醚冲洗产品,离心得到固体后倾倒上清液并用真空烘箱干燥,得到无色透明胶状固体30mg(产率72%),其保存于-20℃冰箱中。以与上述方法类似的方法合成平均聚合度为7的聚l-谷氨酸(乙二醇)3酯,只需将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐投料量改为13mg即可,反应后加入乙酸酐(1.33mg,2.0当量)封端,按照上述方法后处理,maldi-tof质谱表征如图3所示。实施例2:用于蛋白环化的嵌段聚氨基酸p2的合成在手套箱中将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐(n1,13.0mg,0.044mmol,7当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(13.1μl×0.5m,1.0当量),在室温下搅拌25小时后加入甘氨酸-n-羧基内酸酐(n2,2.0mg,0.0197mmol,3当量),继续在室温搅拌2小时,后处理方式与实施例1相同。实施例3:用于蛋白偶联的聚氨基酸p4的合成在手套箱中将磷酸乙酯-l-酪氨酸n-羧基内酸酐(n3,100mg,0.291mmol,20当量)溶解于无水n,n-二甲基甲酰胺(2.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(29.1μl×0.5m,1.0当量),在室温下搅拌25小时,后处理方式与实施例1相同。将上述聚氨基酸(p3,80mg,0.0133mmol,1.0当量)溶解于二氯甲烷(1.5ml)中,并依次加入三乙胺(300μl,2.1mmol)和三甲基溴硅烷(360μl,2.7mmol)。将反应液加热至60℃并搅拌8小时,在旋转蒸发仪上旋干溶剂后将产物溶解于去离子水中(4ml),并加入edta(10mg),用氢氧化钠水溶液(1m)将ph调至中性,并用氯化钠水溶液透析(1000damwco,3次)。溶液冻干后得到淡黄色固体p460mg(产率83%)。实施例4:用于蛋白偶联的聚氨基酸p5的合成在手套箱中将l-谷氨酸-4-丙烯氧基苄酯-n-羧基内酸酐(n4,300mg,0.952mmol,50当量)溶解于无水n,n-二甲基甲酰胺(3.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(38.0μl×0.5m,1.0当量),室温搅拌25小时,后处理方式与实施例1相同。最终得到无色透明胶状固体200mg(产率69%)。实施例5:用于蛋白偶联的聚氨基酸p6的合成将实施例4中制备的聚氨基酸(p5,50mg,0.173mmol,1.0当量按双键的物质的量计算)溶解于n,n-二甲基甲酰胺(2.0ml)中;将巯基乙胺盐酸盐(118mg,1.04mmol,6.0当量)溶解于去离子水中(200μl);混合两份溶液并加入安息香二甲醚(0.89mg,3.46×10-3mmol,0.02当量),摇匀后置于365nm紫外灯下反应3小时。反应结束后将溶液倒入异丙醇中(40ml),离心收集沉淀。真空烘箱干燥后得到无色透明粘稠胶状固体32mg(产率53%)。实施例6:用于蛋白偶联的聚氨基酸p7的合成将实施例1中制备的聚氨基酸(p1,50mg,0.173mmol,1.0当量按双键的物质的量计算)溶解于n,n-二甲基甲酰胺(2.0ml)中;将巯基丙酸(110mg,1.04mmol,6.0当量)溶解于去离子水中(200μl);混合两份溶液并加入安息香二甲醚(0.89mg,3.46×10-3mmol,0.02当量),摇匀后置于365nm紫外灯下反应3小时。后处理方式与实施例5相同,最终得到淡黄色透明胶状固体33mg(产率51%)。实施例7:用于蛋白偶联的聚氨基酸p8的合成在手套箱中将l-谷氨酸-丙烯基(乙二醇)3酯-n-羧基内酸酐(n5,200mg,0.580mmol,50当量)溶解于无水n,n-二甲基甲酰胺(2.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(23.2μl×0.5m,1.0当量),室温搅拌25小时,后处理方式与实施例1相同,最终得到无色透明胶状固体160mg(产率82%)。实施例8:用于蛋白偶联的聚氨基酸的后修饰将实施例7中制备的聚氨基酸(p8,50mg,0.155mmol,1.0当量)按双键的物质的量计算)溶解于n,n-二甲基甲酰胺(2.0ml)中;将巯基乙胺盐酸盐(113mg,0.94mmol,6.0当量)溶解于去离子水中(200μl);混合两份溶液并加入安息香二甲醚(0.84mg,3.30×10-3mmol,0.02当量),摇匀后置于365nm紫外灯下反应6小时。反应结束后将溶液用去离子水稀释至5ml,用脱盐柱pd10分两次处理,冻干后无色透明胶状固体25mg(产率46%)。实施例9:用于蛋白偶联的聚氨基酸p10的合成在手套箱中将n-甲基-n-羧基内酸酐(n6,15.0mg,0.131mmol,20当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(13.6μl×0.5m,1.0当量),室温搅拌25小时,后处理方式与实施例1相同。最终得到白色固体7mg(产率78%)。实施例10:用于蛋白偶联的嵌段聚氨基酸p11的合成在手套箱中将l-苄氧羰基赖氨酸-n-羧基内酸酐(n7,100.3mg,0.328mmol,50当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入三甲基硅基苯硫酚的dmf溶液(13.1μl×0.5m,1.0当量),在室温下搅拌25小时后加入l-谷氨酸苄酯-n-羧基内酸酐(n8,86.1mg,0.328mmol,50当量),继续在室温搅拌25小时,后处理方式与实施例1相同。实施例11:用于蛋白偶联的聚氨基酸p12的合成在手套箱中将l-苄氧羰基赖氨酸-n-羧基内酸酐(n7,100.3mg,0.328mmol,50当量)溶解于无水四氢呋喃(1.0ml)中,并加入三甲基硅基苯硫酚的四氢呋喃溶液(13.1μl×0.5m,1.0当量),在室温下搅拌25小时,后处理方式与实施例1相同。由于在四氢呋喃中引发剂引发效率低,因此实际得到的聚合物聚合度偏大,理论聚合度为300。实施例12:用于蛋白偶联的聚氨基酸p13的合成在手套箱中将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐(n1,13.0mg,0.131mmol,20当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入三甲基锡基苯硒酚的dmf溶液(13.6μl×0.5m,1.0当量),在室温下搅拌25小时,后处理方式与实施例1相同。实施例13:用于蛋白偶联的聚氨基酸p14的合成在手套箱中将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐(n1,13.0mg,0.131mmol,20当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入2-巯基乙烷磺酸钠的dmf溶液(13.6μl×0.5m,1.0当量),在室温下搅拌25小时,后处理方式与实施例1相同。实施例14:用于蛋白偶联的聚氨基酸p15的合成在手套箱中将l-谷氨酸(乙二醇)3酯-n-羧基内酸酐(n1,13.0mg,0.131mmol,20当量)溶解于无水n,n-二甲基甲酰胺(1.0ml)中,并加入巯基丙酸的dmf溶液(13.6μl×0.5m,1.0当量),在室温下搅拌25小时,后处理方式与实施例1相同。在以下表1中示出上述实施例中制备的聚合物的表征参数,并且所示出的实际测量值均是至少三次测量所得的数值的平均值。详言之,所有聚合物的分子量及分子量分布系数(pdi)都由凝胶排阻色谱(gpc)结合9角度激光光散射仪测得,所用流动相为添加0.1m溴化锂的无水n,n-二甲基甲酰胺。表1聚合物分子量及分子量分布聚合物编号理论聚合度实际聚合度实际分子量pdip1202056001.12p210922001.03p3201947001.05p55052173001.08p85049135001.06p10202015001.07p1110099239001.03p12300305670001.14p135050139001.08p145048134001.15p155046130001.06由上表可见,所有聚合物的实际分子量都与理论分子量接近,并且分子量分布很窄,说明聚合过程的可控性优异。实施例15:环状(多肽-聚氨基酸p2(n=7,m=3)偶联物的制备将实施例2中制备的聚氨基酸p2(n=7,m=3)(3.8mg,1.72μmol,2.0当量)与n端带有半胱氨酸的多肽(cys-pep,1.8mg,0.86μmol,1.0当量,序列为cgdakglpetghhhhhhk)溶解于超纯水中,用氢氧化钠水溶液(1.0m)调节ph至7.0,室温反应12小时。经过ninta亲和纯化后,用脱盐柱pd10处理后冻干得到自然化学连接产物白色粉末2.6mg(产率74%)。将自然化学连接产物溶解于含有氯化钠(150mm)和氯化钙(10mm)的tris-hcl缓冲液中(50mm,5.0ml,ph~7.4),并加入转肽酶(sortase)a(150μl×227μm,0.05当量),室温反应0.5小时后,环状多肽-聚氨基酸偶联物的粗产品用ninta提纯(用缓冲液b洗脱,含有20mmtris-hcl,500mm氯化钠,20mm咪唑,ph~8.0)。收集洗脱下来的组分并用脱盐柱pd10处理。冻干后得到白色粉末环状(多肽-聚氨基酸p2(n=7,m=3))偶联物纯品1.6mg,产率82%。产物经过maldi-tof表征,分子量与理论值一致,如图4所示。实施例16:enlyfqc-增强型绿色荧光蛋白-lpetgh6(enlyfq-cys-egfp-lpetgh6)的表达及纯化将n端编码有enlyfq和c端编码有lpetghhhhhh的质粒pet-tev-cys-egfp通过化学转化的方法转入大肠杆菌(e.coli)bl21中。菌种在10ml添加有100μg/ml卡那霉素的lb培养基中复苏10~12小时,然后1:100接种至1l添加有100μg/ml卡那霉素的lb培养基中,在250rpm,37℃的条件下摇至od600=0.8,加入终浓度为1mm的异丙基硫代半乳糖苷诱导细菌表达,培养条件改为250rpm,30℃。5小时后6500rpm,4℃离心40min收集菌体。用20ml的缓冲液a(20mmtris-hcl,500mmnacl,ph8.0)悬菌,在冰浴条件下超声(130w,20khz,50%功率,超声5秒,暂停10秒,超声时间为10min)裂解。裂解液先后经过6500rpm,40min和12000g,40min两步4℃离心,收集上清液,用0.22μm滤膜过滤,经ninta亲和柱纯化,其中,平衡缓冲液为50mmtris,500mmnacl,ph8.0,洗脱液为添加有300mmm咪唑的上述平衡液。最后得到48mg目的蛋白(其序列见序列表中seqidno:1),并经过uplc-ms进行确认(参见图5),计算值为31324,得到31333。分子量误差(9道尔顿)在误差允许范围(10道尔顿)内,因此可确认所得到的蛋白为目的蛋白tev-增强型绿色荧光蛋白-lpetgh6。实施例17:含有1,2-巯基乙胺结构位点特异的增强型绿色荧光蛋白(egfp)的表达步骤1:1,2-巯基乙胺非天然氨基酸的合成将化合物1(233mg,1.00mmol,1.0当量)溶解于无水四氢呋喃中(20ml),加入n,n’-二环己基碳二亚胺(dcc,206mg,1.00mmol,1.0当量)和n-羟基琥珀酰亚胺(nhs,115mg,1.00mmol,1.0当量),在室温下搅拌3小时后过滤除去固体,滤液旋干后,经柱色谱分离,洗脱剂为二氯甲烷∶乙酸乙酯=5∶1(体积比)到纯乙酸乙酯。得到白色固体化合物2(264mg,产率80%)。将化合物2(264mg,0.80mmol,1.0当量)溶解于n,n-二甲基甲酰胺中(20ml),加入boc-lys-oh(246mg,0.80mmol,1.0当量)和三乙胺(0.2ml),室温搅拌3小时后旋干溶剂,经柱色谱分离,洗脱剂为二氯甲烷到二氯甲烷∶甲醇=10∶1(体积比)。得到无色油状液体化合物3(221mg,产率60%)。将化合物3(221mg,0.48mmol,1.0当量)溶解于氯化氢的二氧六环溶液中(10ml),室温搅拌过夜后产品析出,抽虑得到产品,即化合物4(1,2-巯基乙胺非天然氨基酸),其经ph调节后,用于后续实验。步骤2:增强型绿色荧光蛋白的表达将蛋白质基因序列的特定位点突变为tag的质粒(原蛋白序列详见序列表seqidno:2(c端带有his6标签),其第149位的天冬氨酸n对应的密码子突变为tag,得到特定位点突变的基因序列)和携带有琥珀酰氨酰合成酶/trna的质粒通过化学转化的方法同时转入大肠杆菌(e.coli)bl21中。菌种在10ml添加有100μg/ml氨苄青霉素和34μg/ml氯霉素的lb培养基中复苏10~12小时,然后接种至1l添加有100μg/ml氨苄青霉素和34μg/ml氯霉素的lb培养基中,在250rpm,37℃的条件下培养2小时后加入终浓度为2mm的非天然氨基酸4,摇至od600=0.8,加入终浓度为1mm的异丙基硫代半乳糖苷诱导细菌表达,培养条件改为250rpm,30℃。12小时后6500rpm,4℃离心40min收集菌体。用20ml的缓冲液a(20mmtris-hcl,500mmnacl,ph8.0)悬菌,在冰浴条件下超声裂解。裂解液先后经过6500rpm,40min和12000g,40min两步4℃离心,收集上清液,用0.22μm滤膜过滤,ninta纯化方法同实施例16。实施例18enlyfqc-增强型绿色荧光蛋白-lpetgh6(enlyfq-cys-egfp-lpetgh6)的酶切在透析袋(mwco3000)中加入1ml蛋白质溶液(12mg/ml)、5μl烟草蚀纹病毒蛋白酶(0.2mg/ml)和10μl10x缓冲液b(500mm磷酸钠、5mmedta和10mmdtt)。将透析袋置于1l缓冲液b(50mm磷酸钠、0.5mmedta和1mmdtt)中在室温下透析酶切。酶切3小时后得到蛋白cys-egfp-lpetgh6(其氨基酸序列即序列表中seqidno:1去除n端的“menlyfq”后的序列)并经过uplc-ms确认(参见图6),计算值为30398,得到30404。分子量误差(6道尔顿)在误差允许范围(10道尔顿)内,因此可确认所得到的酶切后的蛋白为目的蛋白cys-绿色荧光蛋白-lpetgh6。实施例19:cys-增强型绿色荧光蛋白-lpetgh6与聚氨基酸p1(n=7)的自然化学连接反应(蛋白质n端偶联)1当量的cys-增强型绿色荧光蛋白-lpetgh6与2当量的聚氨基酸p1(n=7)在50mmtris-hcl,2mm三(2-羧乙基)膦的溶液(ph~6.5)中进行自然化学连接反应,反应10h后经过快速蛋白液相色谱(fplc)纯化,得到位点特异的蛋白质高分子杂化材料,并经过变性蛋白质凝胶电泳(sds-pagegel)表征其分子量变化和非变性蛋白质凝胶电泳(nativesds-pagegel)表征其纯度。如图7所示,在sds-pagegel中,聚氨基酸-增强型绿色荧光蛋白偶联物条带相对于原料蛋白质向分子量大处迁移,说明聚氨基酸接枝成功,而变性和非变性凝胶电泳中聚氨基酸-增强型绿色荧光蛋白偶联物都为均一的单条带(二聚体为相同的单体蛋白在天然状态下通过物理相互作用形成的),说明产物纯度较高,其中产率为50%~65%。实施例20:enlyfqc-增强型绿色荧光蛋白-lpetgh6与聚乙二醇-聚甘氨酸在转肽酶介导下的偶联(sortasemediatedligation,蛋白质c端偶联)1当量的enlyfqc-增强型绿色荧光蛋白-lpetgh6(1mg/ml,300μl)、5当量的聚乙二醇-聚甘氨酸(10mm,53μl)和0.1当量的转肽酶(4.9mg/ml,52μl)加入到200μl含有150mmnacl和10mmcacl2的50mmtris-hclph7.4缓冲液中。室温反应30min,经ninta亲和纯化,分离得到产物enlyfqc-增强型绿色荧光蛋白-聚乙二醇-聚甘氨酸偶联物。平衡缓冲液为50mmtris,500mmnacl,ph8.0,上样即开始收集流脱缓冲液,继而用50mmtris,500mmnacl,20mm咪唑,ph8.0洗脱,得到溶解有产物的洗脱液,再用50mmtris,500mmnacl,300mm咪唑,ph8.0的缓冲液清洗树脂,洗脱未反应的蛋白质原料。enlyfqc-增强型绿色荧光蛋白-聚乙二醇-聚甘氨酸偶联物通过变性胶蛋白质凝胶电泳(sds-pagegel)表征其分子量,非变性胶蛋白质凝胶电泳(nativesds-pagegel)表征其纯度。如图8所示,在sds-pagegel中,enlyfqc-增强型绿色荧光蛋白-聚甘氨酸-聚乙二醇偶联物条带相对于原料蛋白质向分子量大处迁移,说明聚氨基酸接枝成功,而在变性和非变性凝胶电泳中聚氨基酸-增强型绿色荧光蛋白偶联物为均一的单条带,说明产物纯度较高,其中产率为56%。实施例21:聚氨基酸p1(n=7)-增强型绿色荧光蛋白-聚甘氨酸-聚乙二醇节点状偶联物的制备以两步骤,依次根据实施例19和20所述的反应和纯化方法,制备聚氨基酸p1(n=7)-增强型绿色荧光蛋白-聚甘氨酸-聚乙二醇节点状偶联物。中间产物聚氨基酸p1(n=7)-增强型绿色荧光蛋白和节点状偶联物均经过变性和非变性蛋白质凝胶电泳表征(参见图9)。产物的分子量逐步向蛋白质分子量高出位移说明高分子先后均接枝成功,且单一条带表明产物纯度较高。两步产率分别为65%和50%。实施例22:增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)-干扰素α哑铃状偶联物的制备1当量的增强型绿色荧光蛋白-lpetgh6(6.5mg)、10当量的聚氨基酸p2(n=7,m=3)(6.6mg)和0.1当量的转肽酶(0.5mg)加入到1ml含有150mm氯化钠和10mm氯化钙的tris-hcl缓冲液(50mm,ph7.5)中,室温反应30min。参照实施例21的纯化方法纯化得到增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)偶联物,产率为55%。继而将增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)偶联物与cys-干扰素α参照实施例21进行自然化学连接得到增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)-干扰素α哑铃状偶联物,产率为50%。中间产物和哑铃状偶联物均经过变性和非变性蛋白质凝胶电泳进行分子量和纯度的表征(参见图10)。偶联物分子量逐步向高分子量处位移,且增强型绿色荧光蛋白-聚氨基酸-干扰素α哑铃状偶联物分子量根据蛋白质分子量标尺分布在43kda~55kda之间,符合预期的增强型绿色荧光蛋白(~31kda)和干扰素α(~21kda)异质双蛋白分子量加和范围(~52kda),因此可确认得到的是增强型绿色荧光蛋白-聚氨基酸-干扰素α异质双蛋白哑铃状偶联物。通过单一条带可确认产物纯度较高。实施例23:环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物的制备参照实施例19利用自然化学连接制备聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白-lpetgh6偶联物。将1当量的聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白-lpetgh6偶联物(0.42mg)与0.1当量的转肽酶混合在含有150mm氯化钠和10mm氯化钙的50mmtris-hcl,ph7.5的缓冲液中,控制聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白-lpetgh6偶联物的终浓度为50mm,室温反应30min,反应结束后参照实施例22进行分离纯化,产率约为42%。利用基质辅助激光解离-飞行时间质量分析器(maldi-tof)、变性胶蛋白质凝胶电泳和免疫印迹分析(westernblot)来表征环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物。聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白-lpetgh6偶联物的碳端比环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物多出7个氨基酸(即,ghhhhhh)。因此利用maldi-tof分析,聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白-lpetgh6偶联物环化后的分子量整体低于环化前(理论是1160da,实际算的~1206da),直接表明增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)形成环状偶联物。此外,变性胶蛋白质凝胶电泳(sds-pagegel)中,环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))由于分子量减小和拓扑结构的变化,条带位置会向分子量低处位移,免疫印迹分析表明组氨酸标签(his-tag)消失,进一步表明成功得到了环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物,具体分析结果参见图11。实施例24:cys-增强型绿色荧光蛋白、聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白、环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物的羧肽酶稳定性测试控制cys-增强型绿色荧光蛋白、聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白和环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物在50mmph7.4的tris-hcl缓冲液中终浓度为4.0mg/ml,羧肽酶的终浓度为80μg/ml,当量比为egfp的0.01当量,37℃孵育。在设定的时间点,取样(5μl)与上样缓冲液混合,95℃煮样10min,终止酶切反应。待最后一个时间点完成取样后,所有样品同时跑胶,利用蛋白质凝胶电泳进行表征。考马斯亮蓝染色和脱色完成后,利用typhoonflalaserscanner对蛋白质进行定量。如图12所示,图a、b、c分别为cys-增强型绿色荧光蛋白、聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白和环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))偶联物在羧肽酶的作用下随时间延长的降解情况。通过typhoonflalaserscanner根据染色后条带的强度对蛋白进行定量,在相同的羧肽酶比例下,野生型增强型绿色荧光蛋白在1小时内基本全部降解,而聚氨基酸p2(n=7,m=3)-增强型绿色荧光蛋白在3小时处只剩20%左右完整的偶联物,相比之下,环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3)偶联物)则在7小时才开始观察到一定程度的降解。因此,相比于线状的拓扑结构,环状(增强型绿色荧光蛋白-聚氨基酸p2(n=7,m=3))在酶耐受性上表现出了极大的优势。实施例25:药物蛋白enlyfqcg-干扰素-lpetgleh6(tev-cys-ifnα-lpetgh6)的表达及纯化将n端编码有menlyfqcg和c端编码lpetglehhhhhh的质粒pet-tev-cys-ifnα-lpetgleh6通过化学转化的方法转入大肠杆菌(e.coli)origamib(de3)中。菌种在10ml添加有100μg/ml氨苄青霉素的lb培养基中复苏10~12小时,然后接种至1l添加有100μg/ml氨苄青霉素的lb培养基中,在250rpm,37℃的条件下摇至od600=0.8,加入终浓度为1.0mm的异丙基硫代半乳糖苷诱导细菌表达,培养条件改为200rpm,30℃。过夜培养后6500rpm,4℃离心30min收集菌体。用20ml的缓冲液a(20mmtris-hcl,500mmnacl,ph8.0)重悬菌体,在冰浴条件下超声裂解。裂解液先后经过6500rpm,40min和12000g,40min两步4℃离心,收集上清液,用0.22μm滤膜过滤,经ninta亲和柱纯化得到98mg目标蛋白,并经过uplc-ms进行确认,计算的理论分子量为21918,得到21914。分子量误差(4道尔顿)在误差允许范围(10道尔顿)内,因此可确认所得到的蛋白为目的蛋白tev-cys-ifnα-lpetgh6。实施例26:药物蛋白enlyfqcg-干扰素-lpetgleh6(tev-cys-ifnα-lpetgleh6)的酶切在透析袋(mwco3000)中加入1ml蛋白质溶液(12mg/ml)、5μl烟草蚀纹病毒蛋白酶(0.2mg/ml)和10μl10x缓冲液b(500mm磷酸钠、5mmedta和10mmdtt)。将透析袋置于1l缓冲液b(50mm磷酸钠、0.5mmedta和1mmdtt)中在室温下透析酶切。酶切3小时后得到蛋白cys-egfp-lpetgh6并经过uplc-ms确认(参见图13),计算值为20991,得到20987。分子量误差(6道尔顿)在误差允许范围(10道尔顿)内,因此可确认所得到的酶切后的蛋白为目的蛋白cys-干扰素α-lpetgh6。实施例27:药物蛋白cys-干扰素-lpetgleh6与不同长度聚氨基酸的自然化学连接反应1当量的目的蛋白cys-ifnα-lpetgleh6与3当量的聚氨基酸phs-p(oeg3-glu)100(l型,p1(n=100))在50mmtris-hcl,2mmdtt的溶液(ph~7.4)中进行自然化学连接反应,反应10小时后经过快速蛋白液相色谱(fplc)纯化,得到位点特异的p(oeg3-l-glu)100-ifnα偶联物。聚氨基酸p1(n=100)-干扰素α偶联物通过15%变性胶蛋白质凝胶电泳(sds-pagegel)表征其分子量和纯度。如图14所示,在sds-pagegel中,聚氨基酸p1(n=100)-干扰素α偶联物条带相对于原料蛋白质向分子量大处迁移,说明聚氨基酸接枝成功,且偶联物为均一的单条带,说明产物纯度较高,其中产率为70%。或者,以与上述相同的方法和条件,进行药物蛋白cys-干扰素α-lpetgleh6与聚氨基酸phs-p(oeg3-glu)100(d,l型氨基酸)和聚氨基酸p2(phs-p(oeg3-glu)n-gly3(n=7或20,m=3))自然化学连接反应,得到的偶联物经fplc纯化后,通过15%变性胶蛋白质凝胶电泳表征。如图14所示,聚氨基酸-干扰素α偶联物条带相对于原料蛋白质均向分子量大处迁移,说明聚氨基酸接枝成功,其中产率为70%~80%。实施例28:cys-干扰素α-lpetg的n,c两端分别与不同长度聚氨基酸的位点特异性偶联采用实施例27中的产物聚氨基酸p1(n=100)-干扰素α-lpetgleh6和聚氨基酸p2(n=20,m=3)在转肽酶的作用下,在20mmtris-hcl,150mmnacl,10mmcacl2的缓冲液中进行转肽酶介导的转肽反应,反应体系经过ninta柱分离纯化得到n,c两端都位点特异偶联聚氨基酸的产物聚氨基酸p1(n=100)-干扰素α-lpet-聚氨基酸p2(n=20,m=3)。产物经过15%变性胶蛋白质凝胶电泳和免疫印迹分析表征。在免疫印迹分析中,如图14所示,由于标签his-tag的离去,产物的抗his标签信号消失表明干扰素α的n,c两均成功接枝聚氨基酸。实施例29:聚氨基酸p2(n=7或20,m=3)-干扰素α-lpetgleh6偶联物的环化反应聚氨基酸p2(n=7或20,m=3)-干扰素α-lpetgleh6偶联物在转肽酶的作用下,在20mmtris-hcl,150mmnacl,10mmcacl2的缓冲液中进行环化反应,经过ninta柱分离纯化得到环状(聚氨基酸p2(n=7或20,m=3)-干扰素α-lpetgleh6)。如图15所示,干扰素α与聚氨基酸的环状拓扑结构的形成可通过sds-page胶中条带相对与环化前的偶联物像分子量低处位移,且其相对应的免疫印迹分析中anti-his信号的消失来确认。实施例30:聚氨基酸p1(n=100)-ifnα与环状(聚氨基酸p2(n=7或20,m=3)-ifnα)的热稳定性测试本实施例中,所采用的目的蛋白为重组人干扰素α,可以通过染料syproorange来测定蛋白质融化温度(tm值),该种染料会结合随温度升高引起的蛋白构象变化而暴露的疏水区,从而荧光发射强度有较大变化。野生型ifnα及各种与聚氨基酸的偶联物的tm值。5μl200×宝石橙蛋白染料(thermo)和45μl(5μm)的待测蛋白混合在1×pbs缓冲液中,共50μl反应体系,加到96孔板中,平行三个孔加样,在荧光定量pcr仪中检测,从37度以2.2℃/min的速度加热到98℃,根据所得曲线计算各蛋白质样品的tm值。如图16所示为各蛋白质样品的tm值,从中可判断各种偶联物的热稳定性,tm值越高则其热稳定性越好。其中聚氨基酸p1(n=100)-干扰素α偶联物和环状(聚氨基酸p2(n=7或20,m=3)-干扰素α)表现出十分优异的热稳定性,尤其环状(聚氨基酸p2(n=7或20,m=3)-干扰素α)的热稳定性相比于目前各文献和专利报道中最好的。实施例31:各种聚氨基酸-干扰素α偶联物的胰蛋白酶耐受性测试本实施例中,以野生型重组人干扰素α为对照,对本公开的聚氨基酸-干扰素α偶联物的胰蛋白酶耐受性进行测试。37℃下,加入0.01当量的胰蛋白酶处理样品,在不同时间点取样检测剩余的完整蛋白比例,结果如图17所示,根据蛋白条带的强度进行定量,得到的数据绘制成曲线图(如图17所示),从曲线图b、c中,可以发现随着偶联物所采用的聚氨基酸的分子量增大,所得到的偶联物对胰蛋白酶的耐受性也随之增强,且根据图a所示,在相近分子量的偶联物中,拓扑结构为环状的偶联物的胰蛋白酶耐受性要明显强于未环化的偶联物。实施例32:野生型干扰素wt-ifnα与聚氨基酸p1(n=100l型或n=100d,l型)-ifnα偶联物的圆二色谱测试本实施例中,使用的聚氨基酸p(oeg3-glu)n(l型)单独具有明显的以α螺旋为主的二级结构,所以将其与ifnα偶联后会对ifnα的二级结构造成一定的影响。配成0.35mg/mlpbs蛋白溶液通过圆二色谱监测二级结构的变化,测试证明,ifnα-聚氨基酸偶联物明显增强了ifnα的二级结构,结果如图18所示。实施例33:野生型干扰素α与各种聚氨基酸-干扰素α偶联物的体外活性测试本实施例中,采用对daudi细胞的生长抑制及其诱导a549细胞抗脑心肌炎病毒(emcv)的活性两方面来分别判断各种聚氨基酸-干扰素α的体外抗肿瘤及抗病毒活性的保留。采用对ifnα敏感的人b淋巴瘤细胞系daudi来作为实验细胞系,检测各种聚氨基酸-干扰素α的体外抗肿瘤活性。具体实验步骤为:5000个细胞/孔铺板至96孔板中,将蛋白质及其与聚氨基酸的偶联物稀释至合适梯度加入到铺有细胞的96孔板中,培养72小时之后,利用cellviabilityassay(普洛麦格)检测细胞活力,利用graphpadprism处理相关数据得到各样品的半数抑制量ic50。实验证明,不同拓扑结构的聚氨基酸-ifnα偶联物抗肿瘤活性有较大不同,具体数据表2所示。表2中,具有二级结构的聚氨基酸与干扰素α偶联后对其活性影响最小,且优于已经在临床应用中的干扰素α-聚乙二醇偶联物佩乐能(peg-intron),环状偶联物次之。由此可见,通过调整干扰素α-聚氨基酸偶联物的二级结构或者拓扑结构可在增强其热稳定及酶耐受性的同时最大程度的保留其活性。本实验通过检测聚氨基酸-干扰素α偶联物刺激后的a549细胞抵抗脑心肌炎病毒(emcv)的活性来判断其体外的抗病毒活性。具体测定步骤为:将a549细胞仪8000~10000个细胞/孔的密度铺板至黑色不透明的96孔板中,24小时后,待细胞完全贴壁,加入梯度稀释于10%fbs,dmem培养基中(10μg/ml,3倍比稀释,共14个浓度点)的聚氨基酸-干扰素偶联物(100μl/孔),与细胞共孵育24小时后,吸走蛋白液,加入一定浓度的emcv病毒液(稀释至2%fbs,dmem培养基中,加入病毒的量须确保未加蛋白的细胞48小时后全部病变),以未加蛋白和病毒的孔为细胞存活100%的对照,以未加蛋白只加病毒的孔为细胞存活0%的对照,48小时后利用celltiterluminescentcellviabilityassay(普洛麦格)检测细胞活力,以判断各种偶联物的抗病毒活性。表2实施例34:wt-ifnα与各聚氨基酸-干扰素α偶联物的血药浓度测定本实施例中,采用sd雌性大鼠作为动物活体实验对象,对sd大鼠进行颈静脉插管手术,恢复48小时后,按150μg/kg的剂量通过静脉插管注射待测样品,每种药物实验数量为3只,分别在1min,15min,30min,1h,3h,6h,9h,12h,24h,48h,72h小时取血。冰上保存半小时后,4000g离心取上清保存于-80℃。血清通过elisa方法检测其中的ifnα含量,各蛋白质样品在血液中的浓度变化曲线如图19所示,其中血药浓度半衰期(t1/2)如表2所示。蛋白聚氨基酸偶联物表现出远高于wt-ifnα2b的血药浓度半衰期,并且偶联有l型聚氨基酸的偶联物效果强(t1/2=9.6h)与已在临床应用的佩乐能几乎一致(t1/2=9.8h),强于与偶联有d,l型聚氨基酸的偶联物(t1/2=7.8h),证明聚氨基酸的二级结构会调整蛋白聚氨基酸偶联物整体的二级结构从而改善其的各种生物学性质与功能。而环状的聚氨基酸-干扰素α偶联物的血液循环时间(t1/2=7.5h)长于其环化前的血液循环时间(t1/2=6.5h),说明聚氨基酸-干扰素α偶联物的拓扑结构对其血液循环同样具有较大影响。实施例35:wt-ifnα,聚氨基酸p2(n=20,m=3)-ifnα偶联物与环状(聚氨基酸p2(n=20,m=3)-ifnα)偶联物的体内抗肿瘤活性本实施例中,以人卵巢癌肿瘤细胞ovcar-3为研究的肿瘤细胞系,在六周龄的balb/c雌性裸鼠体内构建肿瘤模型。将细胞以107/200μl的密度悬浮在1640/基质胶(1:1混合)中,200μl/只裸鼠注射入裸鼠右侧前胸部皮下,三周后,肿瘤大小约为30mm3,将老鼠随机分为四组,每组5只,分别注射pbs,wt-ifnα,聚氨基酸p2(n=20,m=3)-ifnα偶联物与环状(聚氨基酸p2(n=20,m=3)-ifnα)偶联物,剂量为20μg干扰素α/只裸鼠,注射体积为100μl,给药频率为每周一次,每隔3天监测一次肿瘤大小和裸鼠体重。实施例36:cys-人表皮生长因子受体2结合片段(cys-her2fab抗体)的表达及纯化将携带有cys-her2fab(半胱氨酸插入到轻链的n端)抗体的质粒通过化学转化至top10大肠杆菌感受态细胞中,在10mllb培养基中37℃,250rpm活化过夜,按照1:100的比例接种至1ltb培养基中,在37℃,250rpm的条件下摇至od600=0.8左右,加入终浓度为0.2%的阿拉伯糖诱导蛋白表达,30℃,200rpm表达16~20h。表达结束后,6500rpm离心30min,收集菌体。加入150ml裂解液(30mmtris,1mmedta,ph8.0,20%蔗糖),室温下轻轻搅拌半个小时,使菌体均匀分散在裂解液中,加入终浓度为0.2mg/ml的溶菌酶,2.5u/l全能核酸酶,37℃,250rpm摇30min,裂解液先后经过6500rpm和12000g离心各30min,每次都保留上清液。将上清液过0.22μm滤膜,经过proteing树脂纯化,上样后用醋酸钠缓冲液(ph4.5)冲洗4~5个柱体积,50mm,ph2.8甘氨酸溶液(5~8个柱体积)洗脱抗体,得到cys-her2fab抗体。经二硫苏糖醇还原后利用uplc-ms确认蛋白的分子量。理论值抗体重链分子量为24305,轻链分子量为23674。经仪器确认所得抗体重链分子量为24310,轻链分子量为23682,分子量与理论计算值相符合(误差在允许范围内)。因此可确认所得抗体为目标抗体。实施例37:聚氨基酸p1(n=7)与cys-her2fab抗体通过自然化学连接的偶联反应cys-her2fab(5mg/ml,50μl,1.0当量)与聚氨基酸p1(n=7)(30当量)混合,室温反应10~12小时。混合物经过fplc纯化,得到聚氨基酸p1(n=7)-her2fab抗体偶联物,经过蛋白质凝胶电泳表征其纯度和分子量。经二硫苏糖醇还原后抗体的重链和轻链解开,得到分子量约减半的条带,如图21所示。纯化后得到的产物相对于反应前的抗体均向高分子量处位移且还原前条带均一,说明聚氨基酸接枝成功,且产物纯度较高,其中产物产率为26%。由上可见,本公开的蛋白质-高分子杂化材料具有制备工艺简单、可控性高等特点,从而大幅降低蛋白质类药物的修饰成本,并且使其体内性质变得更加稳定,进一步改善血液循环时间并有效地发挥生物学和药理学效应。尽管参考特定的实施例来描述本文所述的实施方案,但是应当理解,本领域技术人员会对其作出多种调整和改变,只要其不违背本公开的范围和主旨即可。sequencelisting<110>北京大学<120>制备蛋白质-聚氨基酸偶联物的方法<130>16c12157cn<160>3<170>patentinversion3.3<210>1<211>275<212>prt<213>人工序列<400>1metgluasnleutyrpheglncysleuasnaspilepheglualagln151015lysileglutrphisglumetvalserlysglyglugluleuphethr202530glyvalvalproileleuvalgluleuaspglyaspvalasnglyhis354045lyspheservalserglygluglygluglyaspalathrtyrglylys505560leuthrleulyspheilecysthrthrglylysleuprovalprotrp65707580prothrleuvalthrthrleuthrtyrglyvalglncyspheserarg859095tyrproasphismetlysglnhisaspphephelysseralametpro100105110gluglytyrvalglngluargthrilephephelysaspaspglyasn115120125tyrlysthrargalagluvallysphegluglyaspthrleuvalasn130135140argilegluleulysglyileaspphelysgluaspglyasnileleu145150155160glyhislysleuglutyrasntyrasnserhisasnvaltyrilemet165170175alaasplysglnlysasnglyilelysvalasnphelysilearghis180185190asnilegluaspglyservalglnleualaasphistyrglnglnasn195200205thrproileglyaspglyprovalleuleuproaspasnhistyrleu210215220serthrglnseralaleuserlysaspproasnglulysargasphis225230235240metvalleuleugluphevalthralaalaglyilethrleuglymet245250255aspgluleutyrlysleuprogluthrglyglyleugluhishishis260265270hishishis275<210>2<211>246<212>prt<213>人工序列<400>2metglylysglyglugluleuphethrglyvalvalproileleuval151015gluleuaspglyaspvalasnglyhislyspheservalserglyglu202530glygluglyaspalathrtyrglylysleuthrleulyspheilecys354045thrthrglylysleuprovalprotrpprothrleuvalthrthrphe505560sertyrglyvalglncyspheserargtyrproasphismetlysarg65707580hisaspphephelysseralametprogluglytyrvalglngluarg859095thrileserphelysaspaspglyasntyrlysthrargalagluval100105110lysphegluglyaspthrleuvalasnargilegluleulysglyile115120125aspphelysgluaspglyasnileleuglyhislysleuglutyrasn130135140tyrasnserhisasnvaltyrilethralaasplysglnlysasngly145150155160ilelysalaasnphelysilearghisasnilegluaspglyserval165170175glnleualaasphistyrglnglnasnthrproileglyaspglypro180185190valleuleuproaspasnhistyrleuserthrglnseralaleuser195200205lysaspproasnglulysargasphismetvalleuleuglupheval210215220thralaalaglyilethrhisglymetaspgluleutyrlysglypro225230235240hishishishishishis245<210>3<211>187<212>prt<213>人工序列<400>3metgluasnleutyrpheglncysglycysaspleuproglnthrhis151015serleuglyserargargthrleumetleuleualaglnmetargarg202530ileserleuphesercysleulysasparghisasppheglyphepro354045glngluglupheglyasnglnpheglnlysalagluthrileproval505560leuhisglumetileglnglnilepheasnleupheserthrlysasp65707580serseralaalatrpaspgluthrleuleuasplysphetyrthrglu859095leutyrglnglnleuasnaspleuglualacysvalileglnglyval100105110glyvalthrgluthrproleumetlysgluaspserileleualaval115120125arglystyrpheglnargilethrleutyrleulysglulyslystyr130135140serprocysalatrpgluvalvalargalagluilemetargserphe145150155160serleuserthrasnleuglngluserleuargserlysgluleupro165170175gluthrglyleugluhishishishishishis180185当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1