一种难溶性药物纳米晶体及其制备方法和应用与流程
本发明属于医药
技术领域:
,涉及难溶性药物纳米晶体及其制备方法和应用。
背景技术:
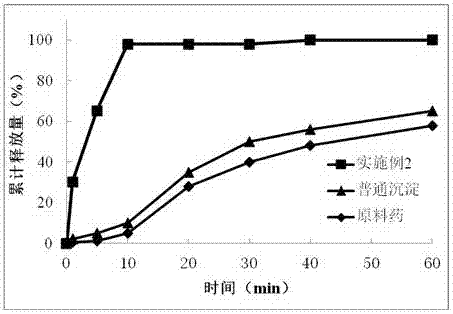
::随着组合化学和高通量筛选等新技术在药物开发领域中的应用,越来越多的有很强药理作用的新化学实体被发现。但是在这些新的候补药物中,水难溶性的化学实体越来越多,这使得很多有着很强药理作用的化学实体难以变成可用于临床治疗的有效药物。有研究报道有约40%的活性化合物由于水溶性差导致它们的生物利用度很低,使它们在开发和应用上受到了很大的限制。如何提高难溶性药物的体内生物利用度一直以来都是制剂学中的一个难题。药物纳米晶体能增加药物溶出度、提高药物的生物利用度、增加药物稳定性及提高药效等,给难溶性药物提供了重现自身价值的机会。微流体技术是在微米尺度调控流体的一项技术,最早用于微量物质的分离和检测,后来也被应用于化学合成、生物分析、光学和信息技术等领域。微流体技术由于其具有独特的层流、表面张力以及毛细管作用等特性,使采用这种方法制备的纳米粒子尺寸更加均匀,同时还具有高通量、批间重现性好、可以实现连续制备等优点。药物纳米晶体为纯药物的纳米尺度的晶体,不含载体,能提高制剂中药物的含量,提高药物传递效率,减小载体材料带来的安全性问题。目前已有报道利用微流体技术制备聚合物纳米粒及用于无机纳米粒子载药的研究。尚无利用微流体技术制备药物纳米晶体,用于提高难溶性药物体外溶出及体内生物利用度的报道。技术实现要素::本发明所要解决的技术问题是提供一种制备难溶性药物纳米晶体的方法及其在药剂学中的应用。本发明通过如下技术方案实现:该方法通过采用微流体控制技术,在微流体装置中控制良溶剂与不良溶剂的微混合实现一步制备难溶性药物的纳米晶体。微混合过程中采用同向流动的良溶剂与不良溶剂,通过调控良溶剂与不良溶剂种类及微混合条件实现对所制备的药物纳米晶体尺寸的调控。良溶剂与不良溶剂为可以互溶,但对药物溶解性差别很大的溶剂,所述的良溶剂选自:乙醇、丙酮、四氢呋喃、二甲基甲酰胺、二甲基亚砜、聚乙二醇。所述的不良溶剂选自:水。进一步地,所述的良溶剂与不良溶剂的组合可以为:水与乙醇,水与丙酮,水与四氢呋喃,水与二甲基甲酰胺,水与二甲基亚砜,水与聚乙二醇。良溶剂与不良溶剂的混合比为5:1—1:40,进一步地,良溶剂与不良溶剂的混合比优选:1:2-1:5。在良溶剂或不良溶剂中可添加适量的表面活性剂或高分子稳定剂。所述的表面性剂为:吐温,司盘,泊洛沙姆,十二烷基硫酸钠等中的一种或几种。所述的高分子稳定剂为:聚乙烯吡咯烷酮,聚乙烯醇,羟丙基甲基纤维素,丙烯酸树脂,壳聚糖,明胶,卡拉胶,环糊精等中的一种或几种。加入表面活性剂的用量为:0-10%,W/W;优选为:0.2-5%。加入高分子稳定剂的用量为:0-10%,W/W;优选为:0.2-5%。具体地,本发明通过如下方法制备:将药物溶于良溶剂中(浓度为1μg/ml-1g/ml);将表面活性剂或稳定剂溶于不良溶剂中;设定良溶剂与不良溶剂的混合比为5:1—1:40之间,进行同向微混合。收集混合液,迅速冷冻干燥,即得相应药物的纳米晶体。所述的药物为难溶性药物。药物在良溶剂中的浓度优选:1mg/ml—200mg/ml。所制备的药物纳米晶体为球状,尺寸在100nm-1000nm之间,能够改变药物在体外的释放速率,可被用于制备口服制剂、注射剂、眼用制剂、外用制剂、吸入制剂及粘膜给药制剂。当在体系中加入适量的表面活性剂及稳定剂的条件下,所制备的纳米晶体尺寸在100nm-500nm之间;进一步地,当混合比控制在1:2-1:5之间时,所制备的纳米晶体尺寸在100nm-200nm之间;目前用于制备药物纳米晶体的方法主要有介质研磨法,高压均质法和沉淀法。其中介质研磨法会利用到研磨介质,介质的磨损残留是该法的一大缺陷,通常不适于注射途径给药及用于治疗慢性疾病的药物。高压均质法制备药物纳米晶体,受药物自身硬度影响很大,同时也存在均质阀损耗导致样品污染的现象。普通的沉淀法是将药物的良溶剂与不良溶剂通过搅拌混合,存在混合不均匀,药物晶体在混合过程中结晶长大、不易控制结晶大小的缺点。本发明通过利用同向微混合方式,实现良溶剂与不良溶剂的层流混合,制备粒径均匀的药物纳米晶体。附图说明:图1为药物纳米晶体的电子显微镜照片。图2为实施例2与沉淀法制备的样品及原料药的溶出曲线。图3为实施例8与沉淀法制备的样品及原料药的溶出曲线。图4为实施例14与沉淀法制备的样品及原料药的溶出曲线。图5为实施例22与沉淀法制备的样品及原料药的溶出曲线。图6为实施例30与沉淀法制备的样品及原料药的溶出曲线。具体实施方式:通过如下实例更详细阐释本发明,应当清楚的是:本发明决不限于实施例,或者仅表现为实施例。实施例1将1g非诺贝特溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得非诺贝特药物纳米晶体。实施例2将1g非诺贝特溶解于10ml丙酮中,作为有机相,以0.5%SDS溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得非诺贝特药物纳米晶体。实施例3将1g洛匹那韦溶解于10ml乙醇中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得洛匹那韦药物纳米晶体。实施例4将1g洛匹那韦溶解于10ml乙醇中,作为有机相,以0.5%PVP溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得洛匹那韦药物纳米晶体。实施例5将0.3g环孢素溶解于10ml乙醇中,作为有机相,以蒸馏水为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得环孢素药物纳米晶体。实施例6将0.3g环孢素溶解于10ml乙醇中,作为有机相,以0.5%SDS溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得环孢素药物纳米晶体。实施例7将0.3g环孢素溶解于10ml乙醇中,作为有机相,以0.2%Poloxamer407溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得环孢素药物纳米晶体。实施例8将0.3g环孢素溶解于10ml乙醇中,作为有机相,以0.2%Poloxamer188溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得环孢素药物纳米晶体。实施例9将1g醋酸地塞米松溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例10将1g醋酸地塞米松溶解于10ml丙酮中,作为有机相,以0.5%SDS溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例11将1g醋酸地塞米松溶解于10ml丙酮中,作为有机相,以0.5%PVP溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例12将1g醋酸地塞米松溶解于10ml丙酮中,作为有机相,以0.2%Tween80溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例13将0.5g塞来昔布溶解于10ml乙醇中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例14将0.5g塞来昔布溶解于10ml乙醇中,作为有机相,以0.2%Poloxamer188溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例15将0.5g塞来昔布溶解于10ml乙醇中,作为有机相,以0.2%Poloxamer407溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例16将0.5g塞来昔布溶解于10ml乙醇中,作为有机相,以0.1%壳聚糖溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得醋酸地塞米松药物纳米晶体。实施例17将1g阿奇霉素溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得阿奇霉素药物纳米晶体。实施例18将1g阿奇霉素溶解于10ml丙酮中,作为有机相,以0.2%Tween80溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得阿奇霉素药物纳米晶体。实施例19将1g阿奇霉素溶解于10ml丙酮中,作为有机相,以0.5%SDS溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得阿奇霉素药物纳米晶体。实施例20将1g阿奇霉素溶解于10ml丙酮中,作为有机相,以0.5%PVP溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得阿奇霉素药物纳米晶体。实施例21将1g尼群地平溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得尼群地平药物纳米晶体。实施例22将1g尼群地平溶解于10ml丙酮中,作为有机相,以0.25%PVA溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得尼群地平药物纳米晶体。实施例23将1g尼群地平溶解于10ml丙酮中,作为有机相,以0.5%PVP溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得尼群地平药物纳米晶体。实施例24将1g尼群地平溶解于10ml丙酮中,作为有机相,以0.2%Poloxamer407溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得尼群地平药物纳米晶体。实施例25将1g硝苯地平溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得硝苯地平药物纳米晶体。实施例26将1g硝苯地平溶解于10ml丙酮中,作为有机相,以0.2%HPMC溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得硝苯地平药物纳米晶体。实施例27将1g西尼地平溶解于10ml丙酮中,作为有机相,以蒸馏水为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得西尼地平药物纳米晶体。实施例28将1g西尼地平溶解于10ml丙酮中,作为有机相,以0.2%的明胶溶液为水相。控制两相混合比为1:10,进行同向层流微混合。反应液冷冻干燥后即得西尼地平药物纳米晶体。实施例29将0.1g羟基喜树碱溶解于10mlDMSO中,作为有机相,以蒸馏水为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得羟基喜树碱药物纳米晶体。实施例30将0.1g羟基喜树碱溶解于10mlDMSO中,作为有机相,以0.2%Poloxamer407溶液为水相。控制两相混合比为1:5,进行同向层流微混合。反应液冷冻干燥后即得羟基喜树碱药物纳米晶体。试验例将制备样品分散于蒸馏水中,利用马尔文激光粒度仪检测样品的粒径,结果见表1。采用普通沉淀法制备的对应于实施例2、8、14、22、30的样品的粒径结果见表2(普通沉淀法制备的样品为将药物溶液直接加入不良溶剂中,搅拌均匀即得)。实施例2、8、14、16、22、30的扫描电子显微镜照片见附图1。实施例2、8、14、22、30与沉淀法制备的样品及原料药的对比溶出曲线见附图2-6(溶出度考察参照中国药典2015版,溶出度测定法(附录XC第二法),转速为100r/min,温度控制在37℃±0.5℃。)。表1各实施例粒径检测结果实施例粒径(nm)实施例粒径(nm)实施例粒径(nm)145311370216352220124302221534331368223468439814261242685656154222531564511655026308733017513273898515182252835294631924629565102312036330221表2采用普通沉淀法制备的样品的粒径结果对应的实施例粒径(μm)235850143022183025当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1