一种氧化还原响应性两亲性共聚物及其制备方法和应用与流程
本发明涉及一种两亲性共聚物及其制备方法和应用,具体说,是涉及一种氧化还原响应性两亲性共聚物及其制备方法和在抗肿瘤领域中的应用,属于药物制剂
技术领域:
。
背景技术:
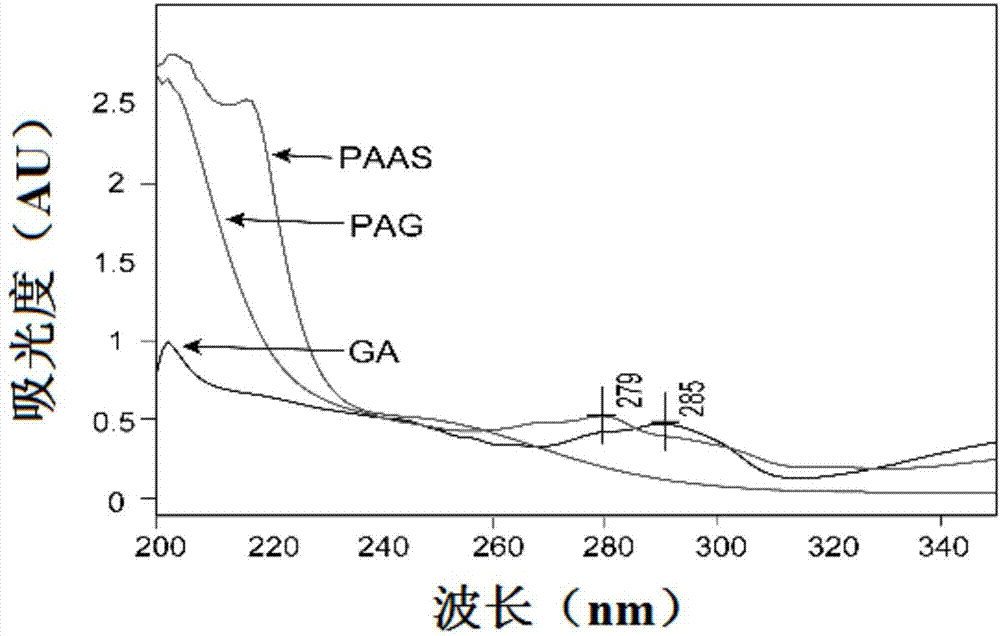
:癌症作为21世纪威胁人类健康的重大疾病之一,具有治愈率低,复发率高等特点,目前已经成为最重要的威胁人类生命健康的疾病之一。据报道,2012年全球范围内癌症共造成820万人死亡,其中以肺癌、肝癌、胃癌、结肠直肠癌为主。过去十年,制药行业中的肿瘤药物供应规模扩大了超过60%,目前有超过500家公司正在从事该领域药物的研发,2015年共有586种抗癌化合物已经进入临床研发后期,十家规模最大的肿瘤药物销售商,有130种候选药物正处在研发后期。目前,治疗肿瘤的方法主要有放射疗法、手术切除法、药物化疗和基因治疗等。肿瘤的发生与生长是多种因素和途径综合作用的结果,上述单一的治疗方法通常只解决一方面的问题,对肿瘤治疗效果有限,经常会伴随肿瘤的多药耐药性产生,例如:手术切除法只能切除显而易见的肿瘤细胞,对于那些微小以及隐匿的癌细胞是无法切除的,这给术后的复发埋下了隐患;而化疗药物的选择性不强,在杀灭癌细胞的同时也会不可避免地损伤人体正常的细胞,从而出现药物的不良反应等。针对此种现象,肿瘤的联合治疗成为一个新兴的研究领域,肿瘤的联合治疗可以通过联合多种治疗手段克服单一治疗存在的问题,通过药物与基因在肿瘤部位作用于不同靶点使得药物之间发挥协同治疗作用。现有的联合治疗主要有各种化疗药物协同治疗、化学药物和基因药物的联合治疗。由于基因与化疗药物的作用机理不同,针对不同肿瘤发生途径,在联合治疗中应用最具前景。在化学药物和基因药物的联合治疗的实际应用中还存在许多问题,主要包括:化疗药物常因水溶性差,导致其在体内生物利用度低;治疗基因表面是带电负性的,由于静电斥力很难进入细胞,即使进入细胞,单独的治疗基因也极易被体液中的核酸酶分解,无法起到治疗作用,必须通过合适的载体将治疗基因包裹着进入细胞,且在恰当的时候将治疗基因释放出来。因此,简单、高效的能共同输送药物和基因的共输送载体是联合治疗成功的关键。两亲性共聚物在水中完全溶解后可自发形成由亲水性外壳和疏水性内核组成的高分子聚合物胶束,并完成对疏水性药物的增溶和包载,亲水性外壳可以保护疏水性药物避免与水相环境接触,并且在体内环境中可以防止被res系统识别,从而增强胶束的稳定性。聚合物胶束具有粒径小,载药量高,可提高水难溶性药物溶解度等特点,这些特点也使得聚合物胶束能够作为理想的抗癌药物递送载体和肿瘤靶向载体。基因载体大体可以分为病毒性基因载体和非病毒性基因载体,阳离子聚合物是近年来发展的一种非病毒性基因载体,如聚精氨酸、聚乙烯亚胺、壳聚糖、聚酰胺-胺和聚赖氨酸等,阳离子聚合物外端含有大量可修饰的基团,在经过功能性分子的修饰后,能够高效率地负载药物和治疗基因,将药物和治疗基因精准地递送到肿瘤细胞内,从而达到抑制肿瘤细胞增殖,杀死肿瘤细胞的作用。尽管与病毒基因载体相比,阳离子聚合物具有可负载任意大小基因、易于制备和低免疫原性等优点,但是转染效率和材料毒性之间的矛盾一直制约着阳离子聚合物作为基因载体的应用,同时具备较低的细胞毒性和较高的转染效率的阳离子聚合物基因载体急需研发。含双硫键的超支化聚酰胺胺外端含有大量活性基团氨基,易于被修饰而使其具有多功能,如接上靶向分子而具有靶向功能或者连接上胶束而具有包载化疗药物的功能。加之其本身呈现正电性,可以负载治疗基因,因此也可以作为基因载体来运用。双硫键的存在赋予材料氧化还原响应性,在肿瘤细胞内高浓度gsh存在下,可以发生双硫键的断裂,从而释放其所包裹的药物或者基因,同时降低材料的毒副作用。目前对超支化聚酰胺胺载体的修饰和改变中,通常是将超支化聚酰胺胺结合一个疏水性的载体基团形成两亲性共聚物用于药物和基因的共同载体,专利cn201410184171.4中公开了一种可降解超支化聚酰胺胺,该专利采用一锅法迈克尔加成聚合反应制备得到可降解超支化聚酰胺胺,在此基础上偶联聚乙二醇(peg)和叶酸(fa),得到叶酸靶向、peg化的可降解超支化聚酰胺胺,得到的聚合物可用作治疗肿瘤的药物载体;李梦艺等人(氧化还原响应性超支化聚酰胺胺衍生物在肿瘤治疗中的应用,《暨南大学》,2016)先用叶酸分子修饰超支化聚酰胺胺,然后通过酰胺化反应在超支化聚酰胺胺上连接聚乙二醇-聚乳酸,合成了聚乙二醇-聚乳酸-超支化聚酰胺胺嵌段共聚物,从而构建一种基因和药物共递送载体。但是上述共聚物对肿瘤的治疗效果有限,还不能满足使用需求。目前还没有采用疏水性的抗癌药物作为疏水嵌段修饰超支化聚酰胺胺形成一种本身具有抗肿瘤作用的高分子药物载体的相关报道,更加没有采用疏水性的抗癌药物作为疏水嵌段修饰超支化聚酰胺胺形成一种本身具有抗肿瘤作用的高分子药物载体,然后该药物载体在水中形成胶束,进而包载第二种抗肿瘤药物,并通过静电作用,还负载一种治疗基因,成为用于治疗肿瘤的药物和基因的共递送载体的相关报道。技术实现要素:针对现有技术存在的上述问题,本发明的目的是提供一种氧化还原响应性两亲性共聚物及其制备方法和在抗肿瘤领域中的应用,以实现基因与化学治疗的联合应用,提高肿瘤的治疗效果。为实现上述发明目的,本发明采用的技术方案如下:一种氧化还原响应性两亲性共聚物,为具有式ⅰ结构的化合物:其中x为带羧基或与乙酸酐衍生化后带羧基的抗肿瘤活性药物。x优先选择藤黄酸(简称ga)、甲氨蝶呤(简称me)或阿霉素(简称dox)中的任意一种。一种制备所述氧化还原响应性两亲性共聚物的方法,包括如下反应:a)式ⅱ化合物(即胱胺)在碱性条件下与丙烯酰氯反应得到式ⅲ化合物(化学名为n,n’-双(丙烯酰)胱胺,简称cba);b)式ⅲ化合物在催化剂存在下与1-(2-胺乙基)哌嗪(简称aepz)进行迈克尔加成反应得到式ⅳ化合物(即超支化聚酰胺胺paas);c)式ⅳ化合物在催化剂和缩合剂存在下与抗肿瘤活性药物x进行酰胺化反应得到式ⅰ化合物(简称为pax);其具体反应路线如下所示:a反应中的碱优选氢氧化钠。a反应中的式ⅱ化合物与丙烯酰氯的摩尔比优选为1:2~1:3。b反应中的催化剂优选氯化钙。b反应中的反应温度优选为45~55℃。b反应中的式ⅲ化合物与1-(2-胺乙基)哌嗪的摩尔比优选为2:3~2:4。c反应中的催化剂优选4-二甲基吡啶(简称dmap),缩合剂优选1-乙基-(3-二甲基氨基丙基)碳二亚胺(简称edc)。c反应中的式ⅳ化合物与抗肿瘤活性药物x的摩尔比优选为2:3~3:2。采用上述技术方案得到的氧化还原响应性两亲性共聚物pax,其分子量为5000~10,0000,将其溶于去离子水后,在去离子水中临界胶束浓度为0.001~0.05mg/ml。一种氧化还原响应性两亲性共聚物在制备抗肿瘤药物中的应用。所述肿瘤包括肺癌、肝癌、乳腺癌、胰腺癌或肠癌。一种所述氧化还原响应性两亲性共聚物自组装形成的胶束,包括所述的氧化还原响应性两亲性共聚物。所述氧化还原响应性两亲性共聚物自组装形成的胶束,为空白胶束或包载有疏水性抗肿瘤活性药物y的载药胶束(该载药胶束简称pax/y)。该自组装形成的胶束其结构中具有抗肿瘤作用的化合物x和双硫键,同时其表面带有正电荷,属于阳离子胶束的一种。所述疏水性抗肿瘤活性药物y选自阿霉素、紫杉醇、多烯紫杉醇、喜树碱、伊立替康、拓扑替康、长春碱、长春地辛中的任意一种。所述载药胶束中,pax与疏水性抗肿瘤活性药物y的质量比优选为优选9:1~99:1,以9:1~19:1为佳。所述氧化还原响应性两亲性共聚物自组装形成的胶束可以通过薄膜挥发法、透析法或乳化法三种方法制备而得,具体如下:采用薄膜挥发法制备氧化还原响应性两亲性共聚物自组装形成的胶束,包括如下操作:将氧化还原响应性两亲性共聚物或氧化还原响应性两亲性共聚物与疏水性抗肿瘤活性药物y溶于极性溶剂中,真空挥发除去极性溶剂,得到空白或载药薄膜,然后加入适量去离子水,室温搅拌,膜过滤,冻干,即得空白胶束或载药胶束。采用透析法制备氧化还原响应性两亲性共聚物自组装形成的胶束,包括如下操作:将氧化还原响应性两亲性共聚物或氧化还原响应性两亲性共聚物与疏水性抗肿瘤活性药物y溶于极性溶剂中,转移至透析袋(mwcutoff=1000)中,置于去离子水中透析,除去极性溶剂,膜过滤,冻干,即得空白胶束或载药胶束。采用乳化法制备氧化还原响应性两亲性共聚物自组装形成的胶束,包括如下操作:将氧化还原响应性两亲性共聚物或氧化还原响应性两亲性共聚物与疏水性抗肿瘤活性药物y溶于极性溶剂中,加入适量去离子水,超声乳化,抽真空除去极性溶剂,膜过滤,冻干,即得空白胶束或载药胶束。采用上述技术方案得到的载药胶束的粒径大小为100-300nm,表面电位为40-70mv。一种氧化还原响应性两亲性共聚物自组装形成的胶束在制备抗肿瘤药物中的应用。所述肿瘤包括肺癌、肝癌、乳腺癌、胰腺癌或肠癌。一种同时负载治疗基因和化疗药物的胶束,将氧化还原响应性两亲性共聚物自组装形成的胶束的溶液与治疗基因z的溶液按照一定的质量比混合,室温孵育,即得所述同时负载治疗基因和化疗药物的胶束(简称pax/y/z)。由于自组装形成的胶束中除了结构中具有抗肿瘤作用的化合物x外,还可用于包载疏水性化疗药物y,并且由于其表面带有正电荷,因此可静电吸附一种治疗基因z,从而形成所述同时负载治疗基因和化疗药物的胶束pax/y/z,该pax/y/z胶束可同时运载两种抗肿瘤药物和一种治疗基因,属于多载药体系,同时结构中双硫键可在肿瘤细胞中高浓度gsh的作用下发生断裂,并释放出抗肿瘤作用的化疗药物x和y,以及治疗基因z,具有氧化应激功能。所述同时负载治疗基因和化疗药物的胶束中,pax与治疗基因z的质量比优选9:1~99:1,以9:1~19:1为佳。所述治疗基因选自p53gene、bcl-2sirna、egfrsirna、vegfsirna中的任意一种。所述负载治疗基因的胶束其粒径为100-300nm,表面电位为-50至+60mv,以0-30mv为最佳值。一种同时负载治疗基因和化疗药物的胶束在制备抗肿瘤药物中的应用。所述肿瘤包括肺癌、肝癌、乳腺癌、胰腺癌或肠癌。总之,由于本发明所述的氧化还原响应性两亲性共聚物pax具有抗肿瘤作用,可以用于制备抗肿瘤药物,因此,基于pax自组装形成的胶束及同时负载治疗基因和化疗药物的胶束均可用于制备抗肿瘤药物。与现有技术相比,本发明具有如下显著性有益效果:本发明的氧化还原响应性两亲性共聚物pax相较于传统的超支化聚酰胺胺衍生物而言,直接由带羧基或与乙酸酐衍生化后带羧基的抗肿瘤活性药物x作为疏水端与亲水的超支化聚酰胺胺paas通过酰胺化反应得到,其主链上含有抗肿瘤活性药物,自身即具有抗肿瘤作用;同时pax是一种具有疏水性和亲水性嵌段的星型嵌段聚合物,由于结构中具有两亲性嵌段,临界胶束浓度较低,可在水中自组装形成胶束,胶束内层疏水端可进一步包裹另一种疏水性抗肿瘤药物y,并且由于胶束外表面带有正电,通过静电作用,还可以进一步负载一种治疗基因z,从而得到一种同时负载治疗基因和化疗药物的胶束,该同时负载治疗基因和化疗药物的胶束作为一种全新的药物和基因的共递送载体,可用于制备抗肿瘤药物,治疗包括肺癌、肝癌、乳腺癌、胰腺癌、肠癌等癌症;由pax得到的胶束pax/y/z,稳定性好,其结构中的双硫键具有氧化应激功能,可在肿瘤细胞中高浓度gsh的作用下发生解离,并释放出所载的药物x、y和基因z,从而实现靶向性,提高药物的生物利用度同时降低毒副作用;另外,本发明制备方法简单,无需特殊设备和苛刻条件,易于实现规模化生产,具有极强的实用价值。附图说明图1为实施例3中pag的核磁氢谱;图2为实施例3中paas、ga、pag的紫外吸收谱图;图3为实施例3中pag的gpc图;图4为实施例3中pag的cmc图;图5a为实施例6中pag-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;图5b为实施例6中pag/dtx-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;图5c为实施例6中dtt存在下pag/dtx-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;图6为实施例7中的细胞转染试验图;图7为实施例8中pag/dtx-mmp-9胶束的透射电镜图;图8为实施例9中pag/dtx的药物释放曲线;图9为实施例10中dtx、dtx-mmp-9、pag/dtx、pag/dtx-mmp-9对mcf-7细胞增殖的抑制曲线(以dtx浓度计算);图10为实施例10中mmp-9、pag-mmp-9、dtx-mmp-9、pag/dtx-mmp-9对mcf-7细胞增殖的抑制曲线(以mmp-9浓度计算);图11为实施例11中nr,pag-nr,mmp-9sirna,pag/nr-mmp-9在mcf-7细胞内吞实验流式结果图;图12为实施例12中pag/icg、icg、空白裸鼠小动物成像实验结果;图13为实施例13中pag/dtx-mmp-9、pag-mmp-9、抗mcf-7乳腺癌的药效结果;具体的13a为mcf-7荷瘤裸鼠肿瘤生长情况;13b为荷瘤裸鼠体重变化;13c为荷瘤裸鼠肿瘤重量比较;13d为荷瘤裸鼠肿瘤图片;与模型组比较,*p<0.05,**p<0.01;图14为实施例13中mcf-7荷瘤裸鼠心、肝、脾、肺、肾、肿瘤h&e组织切片图。具体实施方式下面结合具体实施例进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。实施例1:化合物ⅲ(n,n’-双(丙烯酰)胱胺)的制备:将2.5g胱胺盐酸盐(化合物ⅱ)、1.8gnaoh溶于15.5ml去离子水中,冰浴条件下,滴加丙烯酰氯的二氯甲烷溶液(3.3ml丙烯酰氯溶解于3.3ml无水二氯甲烷),滴加完毕撤去冰浴,室温下反应6小时,结束反应,反应液加入200ml二氯甲烷萃取,萃取三次,合并有机相,有机相经减压蒸馏和真空干燥得白色粉末状物质,即化合物ⅲ(n,n’-双(丙烯酰)胱胺)。经测试:1hnmr(600mhz,cdcl3):δ6.68(s,1h),6.34(dd,j=17.0,1.5hz,1h),6.24(dd,j=17.0,10.2hz,1h),5.69(dd,j=10.2,1.5hz,1h),3.69(q,j=6.4hz,2h),2.90(t,j=6.5hz,2h)。实施例2:化合物ⅳ(超支化聚酰胺胺paas)的制备:将0.642g化合物ⅲ和0.592g无水氯化钙溶于15ml甲醇水溶液中(甲醇/水的体积比为3/1)中,氮气保护下,反应液升温至50℃,搅拌下滴加0.16ml1-(2-胺乙基)哌嗪(aepz),滴加完毕后,于50℃保温反应36小时,然后再次滴加0.35ml1-(2-胺乙基)哌嗪(aepz),继续保温反应4小时,结束反应,加入稀盐酸将反应体系ph调节至3-4,然后将反应液加入透析袋(mwcutoff=1000)中,透析24小时,离心去除沉淀,溶液冷冻干燥得淡黄色粘稠物,即化合物ⅳ(超支化聚酰胺胺paas)。经测试:1hnmr(600mhz,d2o)δ3.60(m,4h,conhch2ch2s),3.30(m,4h,nch2ch2co),3.25(m,4h,nch2ch2co),2.92(m,4h,conhch2ch2s),2.5-2.9(m,12h,ch2fromaepz)。实施例3:氧化还原响应性两亲性超支化聚酰胺胺-藤黄酸共聚物pag的制备:将0.5g藤黄酸(ga)和0.1g4-二甲基吡啶(dmap)溶于10ml无水二氯甲烷中,在冰浴条件下,加入0.3g1-乙基-(3-二甲基氨基丙基)碳二亚胺(edc),冰浴搅拌1小时,然后加入1.5g化合物ⅳ,室温反应24小时,反应结束后,将反应液加入至透析袋(mwcutoff=3500)中,透析24小时,离心去除沉淀,溶液冷冻干燥得黄色粘稠物,即超支化聚酰胺胺-藤黄酸共聚物(简称pag)。图1为pag的核磁谱图(1h-nmr(dmso-d)),图中δ5.0-9.0ppm属于藤黄酸的特征峰(h),同时cba和1-(2-胺乙基)哌嗪的特征峰(a-f)也在氢谱中标出,证明制得的化合物为所需要的超支化聚酰胺胺-藤黄酸共聚物pag。图2为ga、paas和pag的uv谱图;从图中可见,在200nm,paas有末端吸收,而藤黄酸吸收较小,其结合物pag保留了200nm处的末端吸收,同时,藤黄酸在285nm处有吸收峰,paas在此处吸收峰较小,pag此处的最大吸收峰红移至279nm,提示所制备的pag具有paas与ga共同的紫外光谱特征;同时根据藤黄酸在279nm处的吸光度标准工作曲线,测定pag在波长279nm处的吸光度,pag中藤黄酸的含量在16.1-23.2%范围内,证明制得的化合物为所需要的超支化聚酰胺胺-藤黄酸共聚物pag。对制得的超支化聚酰胺胺-藤黄酸共聚物pag进行性能测试,具体如下:a)pag的氧化还原响应性测试:采用凝胶渗透色谱仪测定pag的分子量以及其在相同作用时间的还原剂gsh环境下,pag的分子量大小变化,具体为:将实验分为两组进行,a组:无gsh处理组;b组:100mmgsh水溶液;两组同时加入pag,室温下搅拌4h,搅拌结束后,将样品冻干,重新溶解在dmf(二甲基甲酰胺)中,pag样品浓度为2mg/ml,利用凝胶渗透色谱仪对两组溶液进行分析;实验仪器:agilenthplc1260-示差检测器,流动相:dmf,柱温:35℃,流速:1.0ml/min,并且记录保存凝胶渗透色谱图;以相同色谱条件下采用一系列不同分子量的聚甲基丙烯酸甲酯标准样品的色谱峰峰位作标准曲线,计算样品分子量及分子量分布情况。具体实验结果如图3所示。图3为pag的gpc图;上面的谱图为a组,a组在没有gsh作用下,pag分子量为单峰,分子量分布系数为1.65,分子量mw为1.542*104;下面的谱图的b组,b组在10mmgsh作用4h后,出峰时间右移至4.2min,分子量mn为1879,分子量明显的减小,表明100mmgsh作用4h后pag发生降解,大分子链断裂成小分子片段;从结果可以看出,当100mmgsh作用4h后pag就会发生降解,而肿瘤细胞内gsh的浓度远大于10mmgsh,故在肿瘤细胞内pag会发生降解,从而说明本实施例的pag具有氧化还原响应性。b)pag的临界胶束浓度测定:采用经典的芘荧光探针法对共聚物pag临界胶束浓度值(cmc)进行测定,测定方法如下:1)配制芘浓度为5×10-5mol/l的丙酮溶液,分别将此母液100μl转移至一系列的10ml棕色容量瓶中;2)取适量pag,用水配置至浓度为3mg/ml水溶液,吸取不同体积pag溶液加入到上述含芘的棕色容量瓶中,用去离子水稀释至刻度,得到荧光探针芘终浓度为5×10-7mol/l,最终配成pag浓度分别为1×10-4、5×10-4、1×10-3、5×10-3、1×10-2、2×10-2、3×10-2、4×10-2、6×10-2、8×10-2、0.1、0.2、0.4、0.6mg/ml的一系列浓度的混合溶液;3)将这些混合溶液置于180w功率下水浴超声40min,40℃水浴过夜使其充分平衡;4)使用荧光分光光度计测量其荧光光谱,荧光扫描的激发波长为334nm,发射及激发狭缝宽度分别是2.5和5.0nm,将扫描速度设为240nm/min,记录芘在不同浓度pag溶液中的荧光光谱图,以pag的浓度为横坐标,i1(373nm)/i3(384nm)为纵坐标,绘制不同浓度下芘的i1/i3强度比的变化曲线。芘荧光探针法就是利用该特点,将低溶解度的芘溶解在两亲性聚合物pag溶液中,通过芘溶解度的变化来测定聚合物的cmc值。当两亲性共聚物的浓度超过cmc并形成胶束时,由于其内部疏水基的增溶作用,芘的溶解度急剧增加,导致芘在水溶液中i1/i3降低,据此,可以准确的测定聚合物的cmc值。具体测定结果如图4所示。图4为pag的cmc图,从图中可见在c(pag)为0.03mg/ml时i334/i337荧光强度的比值曲线有1个明显的拐点,可以确定pag对应的临界胶束浓度为0.03mg/ml,临界胶束浓度较低,便于在水中自组装形成胶束进一步包载抗肿瘤药物或治疗基因。实施例4:pag/dtx载药胶束的制备:将pag共聚物溶于甲醇,将多烯紫杉醇(dtx)用甲醇溶解成1mg/ml,按照不同比例(pag与多烯紫杉醇的质量比为1:1、2:1、4:1、9:1、19:1、49:1、99:1)将两者涡旋混匀后,通过旋转蒸发仪,在40℃,100rpm的条件下,以-0.09mpa的真空度缓慢旋干甲醇,形成载药薄膜,然后将载药薄膜置于真空干燥器内,抽真空干燥3小时,除去残留的甲醇,室温条件下以2ml去离子水磁力搅拌水化4h,然后2000r离心5min除去没有包封的dtx,用0.45μm醋酸纤维素酯膜过滤,滤液冻干,即得pag/dtx载药胶束冻干粉,每组平行实验3份。分别测定各组pag/dtx载药胶束中多烯紫杉醇(dtx)的载药量、包封率、粒径、zeta电位,具体如下:a)载药量和包封率的测定:称取适量冻干pag/dtx载药胶束于10ml容量瓶中,加入甲醇溶解并定容得到待测样品。利用hplc测定样品中多烯紫杉醇的浓度及质量,根据以下公式计算载药量及包封率:载药量(lc%)=胶束内的多烯紫杉醇质量/胶束的质量*100%;包封率(ee%)=胶束内的多烯紫杉醇的质量/药物投入量*100%;具体测试结果如表1所示。b)粒径及粒径分布、zeta电位测定:取适量冻干pag/dtx载药胶束,用去离子水配成浓度为0.5mg/ml的样品,利用马尔文纳米粒度、zeta电位和绝对分子量分析仪测定载药胶束的粒径及其分布,检测角度为90°,测试温度为25℃;具体测试结果如表1所示。表1不同比例pag/dtx载药胶束的性能测试数据pag/dtx载药率包封率size电位1:117.4%±2.3%7.2%±1.1%215.1±34.6nm44.8±8.9mv2:117.7%±2.4%26.7%±4.3%257.5±18.7nm44.3±0.7mv4:112.4%±1.3%39.2%±2.9%204.4±27.4nm50.8±3.6mv9:18.5%±1.0%92.2%±5.7%145.7±10.4nm60.2±6.2mv19:16.4%±0.6%87.9%±3.3%115.9±13.7nm63.4±2.6mv49:13.4%±0.3%88.6%±3.3%117.7±15.2nm58.5±3.6mv99:10.5%±0.1%94.3%±1.9%109.0±7.2nm55.1±2.2mv由表1可见:pag水溶液胶束在包载不同剂量的dtx时,包封率随着载药率的增加而迅速降低,兼顾载药率与包封率实验结果,将选择pag:dtx质量比为9:1为最佳投料比,使得dtx的包封率高达92.2%,在此比例下,本申请的pag水溶液胶束可以很好的负载疏水性抗肿瘤活性药物dtx,显著增加疏水性抗肿瘤药物的溶解度,生物利用度好;载药胶束的表面电位随dtx载药量的变化,影响不大,在44.8-63.4mv的范围内,呈电正性,使得其所带的表面正电荷有利于基因的运载,为其进一步包载治疗基因提供了理论依据;制备的载药胶束粒径为纳米级别。实施例5:包载治疗基因的pag/dtx/mmp-9胶束的制备:取pag/dtx(质量比为9:1)胶束冻干粉(含2mgpag计),配置成1mg/mlpag/dtx的水溶液,经孔径为0.45μm的微孔过滤头处理后,把pag/dtx与mmp-9(全称mmp-9-sirna,是sirna的一种)溶液分别按照一定质量比(1:1、2:1、4:1、9:1、19:1、49:1、99:1)进行静置混合,室温孵育30分钟,即可得到pag/dtx-mmp-9胶束,测定各pag/dtx/mmp-9胶束的粒径及粒径分布、zeta电位,测定结果如表2所示。表2pag/dtx/mmp-9胶束的性能测试数据投料比(质量比)是否有dtx析出size电位1:1沉淀221.4±8.4nm-46.3±3.2mv2:1沉淀233.7±1.2nm-29.8±0.1mv4:1浑浊411.0±38.0nm-13.4±0.6mv9:1乳光238.6±23.7nm5.6±2.1mv19:1澄清170.4±1.2nm22.7±1.1mv49:1澄清162.4±2.6nm44.2±0.8mv99:1澄清171.6±6.354.9±1.3mv由表2可见:由于超支化聚酰胺胺含有大量的氨基,故pag电位呈现正电性,而mmp-9是带负电,由于细胞膜表面带负电,载体与基因所形成的复合物想要进入细胞,呈正电更有利于药物的吸收;zeta电位分析结果表明pag/dtx与mmp-9能形成带正电性的胶束,这为其作为基因载体运载基因进入细胞提供可能性,pag/dtx-mmp-9胶束的zeta电位随着mmp-9加入比例的增加,电位逐渐减小,并且由正电荷转为负电荷,当pag-mmp-9质量比为4时,pag/dtx-mmp-9胶束的表面电位由正电转为负电;胶束的粒径随着mmp-9加入比例的增加逐渐增大,直至出现沉淀,根据文献报道,粒径在50-200nm之间的粒子容易进入细胞,兼顾考虑所形成pag/dtx-mmp-9胶束的电位和粒径,当pag:mmp-9的质量比范围在9:1-19:1之间时,所形成电位在22.7至44.2mv之间,纳米粒大小在170nm,符合临床治疗要求,因此,pag-mmp-9的质量比范围为9:1-19:1制备得到的胶束符合临床治疗要求。实施例6:凝胶电泳实验采用凝胶电泳实验来评价pag对mmp-9的结合能力,具体如下:电泳凝胶板由1%的琼脂糖(含0.2μg/ml的溴化乙锭)制得,分别取10μl的聚合物/基因复合物注入凝胶板上的凹槽内,用1×tae做电泳缓冲液,140伏电压下电泳15分钟;然后通过biodoc-ittmsystem凝胶成像系统将电泳结果成像;分别考察pag/dtx(9:1)和mmp-9不同质量比(0/4/9/14/19)时,形成pag-mmp-9胶束的稳定性,且考察pag/dtx(9:1)-mmp-9胶束的稳定性;同时另一组样品加入还原剂dtt(二硫苏糖醇),使之终浓度为3mm,考察dtt对pag/dtx(9:1)-mmp-9胶束的稳定性的影响;具体结果如图5a-c所示。图5a为pag-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;图5b为pag/dtx-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;图5c为dtt存在下pag/dtx-mmp-9在不同质量比下的琼脂糖凝胶电泳分析图;从图5a可知,当pag和mmp-9质量比为0、4时,mmp-9可从电场中迁出,表明pag不能与mmp-9形成稳定的复合物;直到两者质量比为9及以上时,显示mmp-9不能从电场中迁移出,表明pag和mmp-9已形成稳定的复合物;而相同条件下,pag和mmp-9质量比为9时,pag/dtx-mmp-9仍能观察到mmp-9能从电场中迁移出(见图5b),从而可以推测dtx的载入后,在一定程度会影响pag对mmp-9的复合作用,直到两者质量比为14及以上时,显示mmp-9不能从电场中迁移出;因此,选择以pag/mmp-9=14:1为本实验进一步研究的处方;同时在3mmdtt作用下(见图5c),pag结构中的双硫键发生解离,使得形成胶束结构破坏,并进一步破坏pag/dtx(9:1)-mmp-9的稳定性,使得pag/dtx(9:1)和mmp-9质量比>14时,mmp-9sirna均能从电场中迁移出,之前形成的稳定胶束在强还原剂环境中发生解离。该实验结果提示本申请的pag在肿瘤细胞中还原性条件下,可以发生定位解离,释放出抗肿瘤药物与基因药物,发挥其抗肿瘤作用。实施例7:细胞转染实验本实验使用的mmp-9sirna是进入细胞可表达绿色荧光蛋白的基因,从大肠杆菌中提取。取对数期生长的mcf-7细胞,调整细胞浓度以每孔105个接种于24孔细胞培养板中,在37℃、5%co2条件下培养至细胞饱和度为70%;接着换(1x)无血清细胞培养基孵育2小时,同时将一定体积的含pag-mmp-9和pag/dtx-mmp-9的无血清或有血清培养液铺加到培养皿中,确保每孔中mmp-9sirna的含量为1μg。以pei-25k/mmp-9作为阳性对照组,裸mmp-9作为阴性对照组,然后将24孔板置于培养箱中,在37℃、5%co2的环境下培养4小时后,将培养液换掉,加入含血清的培养液继续培养48小时。在荧光显微镜下定性研究绿色荧光蛋白的表达情况,然后吸出培养液,加入一定量的胰酶消化细胞并加入一定的培养基终止胰酶作用,细胞样品经离心后重新悬浮于pbs中,采用流式细胞仪定量分析细胞转染个数,即样品的基因转染效率。为了定量分析pag的转染效率,本项目进行了在无血清条件下和有血清条件下实验,pag和pag/dtx分别与mmp-9形成的胶束的转染试验,试验中依据凝胶电泳试验结果,设定pag/dtx的质量比为9:1,pag与mmp-9质量比为4、9和14,对照组为pei-25k与mmp-9质量比为1.3:1时所形成的胶束,在此时pei-25k能达到最好的转染效率;具体结果如图6所示。图6为细胞转染试验图,其中6a、6b分别为无血清和有血清条件下mcf-7细胞转染结果图;6c、6d分别为在有无血清条件下细胞转染的荧光照片;从图6可见,在无血清的实验条件下,与pei相比,pag和pag/dtx均表现出较好的转染效率,pag与mmp-9质量比为9时,转染效率为44.9%±3.9%,高于pei的转染效率39.1%±1.0%;且当pag与mmp-9质量比为14时,可达到pag最好的转染效率,约48.5%±3.8%;随着dtx的载入,对于pag材料承载mmp-9的转染效率有一定影响;当pag与mmp-9质量比为4时,dtx的载入使得pag对mmp-9的转染效率下降了56%;随着pag比例的增加,其影响逐渐缩小,分别降为18.1%和7.6%;与此同时,我们发现血清的存在对pei、pag对mmp-9的转染效率均有不同程度的影响,与无血清的转染效率相比,pei在有血清下的转染效率下降了32.0%;而当pag与mmp-9质量比为14时,血清对pag的转染效率影响较小,转染效率依然高达49.6%±2.8%,而在dtx存在的情况下,pag有血清下的转染效率下降了24.8%,仍高达33.7%±4.2%。综上所述,pag对mmp-9的转染效率优于pei,说明本实施例中的pag对mmp-9基因具有较好的转染效率。实施例8:验证性实验按照pag/dtx-mmp-9质量比为9:1:0.64进行三批胶束样品的平行制备,并分别测定各组胶束中多烯紫杉醇的载药量、包封率、粒径及粒径分布、zeta电位,具体测试结果如表3所示;同时将平行制备的pag/dtx-mmp-9胶束分别用去离子水配置成相同浓度的pag/dtx-mmp-9胶束溶液,并滴到表面附有支撑膜的铜网上,用滤纸吸去多余的水,用1%磷钨酸进行负染,待样品37℃晾干后,在加速电压为80k下利用透射电子显微镜观察胶束的形态,具体测试结果如图7所示。表3验证性实验测试数据批次载药率包封率粒径(nm)pdi电位(mv)018.5%92.3%170.00.17222.7028.7%93.6%171.30.18123.5038.7%93.5%175.10.17521.6由表3可见:平行三批次的试验结果基本稳定,pag/dtx-mmp-9胶束的粒径大小稳定在170nm左右,电位稳定在20mv。图7为本实施例中ag/dtx-mmp-9胶束的透射电镜图,从图中可见载药胶束的形态为球形或类球形,形貌结构好。实施例9:体外药物释放实验称取冻干的pag/dtx胶束约10mg,配置成含有2.5mg/mlpag的水溶液,分别取0.8ml胶束溶液用含有3mmdtt的pbs将胶束溶液稀释2倍,将混合液装入截留分子量为6kd-8kd的透析袋中,然后将透析袋分别置于50ml含还原剂dtt浓度为3mm、(模拟细胞外基质、血液及肿瘤部位还原性物质的浓度)的pbs(ph7.4)中;上述溶液于37℃下恒温震荡,转速为100rpm,分别于0,0.5、1、2、4、6、8、12、24h时间点取0.1ml透析袋内液,采用高效液相色谱仪测定释放液中不同时间点的多烯紫杉醇dtx的浓度,采用紫外分光光度法测定藤黄酸ga的含量,绘制多烯紫杉醇和藤黄酸的释放曲线;每个时间点每个样品设置三个平行样,具体测试结果如图8所示。图8为本实施例中pag/dtx的药物释放曲线;从图中可以看出,在没有还原剂dtt的作用下,pag/dtx胶束粒子缓慢释放出dtx和ga,在释放24小时后,dtx的总累积释放百分率低于60%,ga的总累计释放百分率不超过20%,这表明pag载药胶束在正常血液循环中有较好的药物包裹能力,不会出现较大的药物泄露;而在还原剂条件下,由于双硫键敏感的还原响应性,载药粒子在24小时内dtx和ga的累积释放率大大增加,在3mmdtt释放介质中,pag/dtx胶束粒子中dtx和ga的释放率分别为80.0%±2.0%,80.0%±6.0%。以上实验结果表明,pag共聚物自组装形成的pag前药胶束能在肿瘤细胞内高还原性环境下,释放出ga发挥抗肿瘤作用,同时也能作为药物载体,负载dtx类难溶性药物至肿瘤及其他病患部位。实施例10:体外毒性实验采用celltiter-gloluminescentcellvaiabilityassay法评价dtx、mmp-9、dtx-mmp-9、pag/dtx、pag-mmp-9或pag/dtx-mmp-9对mcf-7乳腺癌细胞增殖的抑制作用。具体实验过程如下:使用的培养基为含有10%fbs和1%双抗(青链霉素混合液)的rpmi1640培养基,细胞培养环境为37℃及5%co2,细胞每2天换液一次,每4天传代一次;取对数期生长的mcf-7细胞,加入10%fbs的rpmi1640培养基中,调整细胞悬液浓度,然后每孔加入100μl细胞悬液于96孔细胞培养板,铺板使待测细胞调密度至8000个/孔(边缘孔用无菌pbs填充,以防止水分的散失);在37℃、5%co2条件下孵育12小时,使细胞贴壁呈单层生长;吸去培养液后,加入新鲜的含有不同浓度的dtx、mmp-9、dtx-mmp-9、pag/dtx、pag-mmp-9或pag/dtx-mmp-9的1%dmso培养液在37℃培养48h,每个样品设置6个复孔,设置3个只有含有1%dmso培养基的复孔作为空白,3个加含有1%dmso培养基不含药物培养的mcf-7作为对照组;孵育结束后,弃去旧的培养基,每孔加入含有100μlcelltiter-glo试剂,室温再继续培养10min,200r震摇10min,转移至四面不透光的96孔板,使用多功能酶标仪检测化学发光值(实验重复操作三次),计算细胞活性,具体实验结果如图9、图10以及表4、表5所示。图9为dtx,dtx-mmp-9,pag/dtx,pag/dtx-mmp-9对mcf-7细胞增殖的抑制曲线(以dtx浓度计算);从图9中可见,通过制备pag/dtx和pag/dtx-dna胶束,能够增强dtx对mcf-7的抑制作用。图10为mmp-9,pag-mmp-9,dtx-mmp-9,pag/dtx-mmp-9对mcf-7细胞增殖的抑制曲线(以mmp-9浓度计算);从图10可见,mmp-9对mcf-7细胞的增殖基本无抑制作用,而通过制备pag-mmp-9,和pag/dtx-mmp-9胶束,能增强mmp-9对mcf-7的抑制作用。实验结果表明,dtx、pag/dtx、pag-mmp-9、pag/dtx-mmp-9对mcf-7细胞的增殖均有一定程度的抑制作用,如图12、13所示。其中图12显示,通过制备pag/dtx和pag/dtx-dna胶束,能够增强dtx对mcf-7的抑制作用。表4dtx,dtx-mmp-9,pag/dtx,pag/dtx-mmp-9对mcf-7细胞增殖的抑制数据(以dtx浓度计算)μmdtxdtx-mmp-9pag-dtxpag-dtx-mmp-9ic502.394±0.6471.916±0.2211.480±0.250*1.047±0.145**▲▲注:**与dtx组的比较,p<0.01;*与dtx组的比较,p<0.05;▲▲与pag-dtx组的比较,p<0.01;*与dtx组的比较,p<0.05。由表4可见:dtx-mmp-9相较于dtx,对mcf-7的抑制作用物显著性增强,而pag/dtx和pag/dtx-mmp-9相较于dtx,其ic50分别减少了38.2%和56.3%,对mcf-7的抑制作用物显著性增强。表5mmp-9,pag-mmp-9,dtx-mmp-9,pag/dtx-mmp-9对mcf-7细胞增殖的抑制数据(以mmp-9浓度计算)ug/mlmmp-9pag-mmp-9dtx-mmp-9pag/dtx-mmp-9ic50∞15.007±0.6463.169±0.538**2.274±0.315**注:**与pag-dna组的比较,p<0.01。由表5可见:由于mmp-9质粒由于无法进入肿瘤细胞而发挥作用,因此单纯的mmp-9对mcf-7肿瘤细胞的增殖几乎无抑制作用,而制备pag-mmp-9胶束可发挥mmp-9对mcf-7乳腺癌细胞的抑制作用,同时通过制备pag/dtx-mmp-9,可进一步将pag-mmp-9的ic50降低了84.8%,且pag同时载dtx和mmp-9体系,抑制作用最强,可见pag是运载mmp-9质粒对于mmp-9质粒的细胞毒性发挥的关键因素。综上可见,pag可以显著增强dtx和mmp-9对mcf-7乳腺癌细胞增殖的抑制作用,pag同时载dtx和mmp-9体系,抑制作用最强,从而说明pag有望开发为用于治疗肿瘤的药物和基因的共递送载体。实施例11:细胞内吞实验11.1激光共聚焦显微镜实验为了探索pag/dtx-mmp-9胶束在mcf-7细胞中的内吞情况,我们选择与dtx脂溶性相似的细胞膜荧光探针尼罗红(nilered,nr)代替dtx作为模型药物,并选择荧光fam-sirna代替治疗基因mmp-9,制备pag/nr-fam-sirna胶束粒子,探索pag胶束粒子在mcf-7细胞中的分布,同时用4’,6’-二眯基-2-苯基吲哚(dapi)对细胞核染色(dapi在405nm激发波长下发出强烈的蓝光),通过激光共聚焦在nr通道、dapi以及fam-sirna通道下观察pag/nr-sirna胶束在细胞中的内吞以及降解情况,从而探索pag/dtx-mmp-9进入细胞作用的过程。实验过程:培养的mcf-7细胞复苏后传代一次,培养条件为rpmi1640培养基添加10%的胎牛血清、青霉素(100μ/ml)与链霉素(100μ/ml);将培养好的mcf-7细胞铺于激光共聚焦培养皿,每孔2×104个/孔;培养24h后将准备好的pag/nr、nr、pag/nr-fam-sirna、fam-sirna等溶液100μl分别加入培养皿中孵育1、4小时;细胞用预冷的pbs清洗两次(终止内吞作用);加入预冷的200μl、4%多聚甲醛,室温下固定30min,pbs清洗三次;细胞核染色:将4’,6’-二眯基-2-苯基吲哚(dapi)稀释为1μg/ml,每孔加入100μl,室温下染色3min,pbs洗三次;加入500μlpbs,共聚焦激光显微镜下观察,其中nr的激发波长为488nm,dapi的激发波长为405nm,fam-sirna的激发波长为552nm。实验结果如下:mcf-7细胞孵育1h后,便可在pag/nr-fam-sirna作用的mcf-7细胞基质中观察到nr发出的红色荧光,且该荧光强度明显高于nr组,pag/nr组与pag/nr-fam-sirna荧光强度相当,并于细胞核的蓝色荧光有着较强的共定位,说明pag胶束可以明显提高mcf-7细胞对纯nr的内吞效果,而且作用时间较快,也侧面说明pag胶束可以提高mcf-7细胞对所载药物的吸收;同时实验结果显示单独的fam-sirna作用于mcf-7细胞,无法进入细胞内部,但经pag运载的fam-sirna能迅速进入细胞内,可观察到绿色荧光,而且这种作用不受pag对dtx的运载干扰,说明pag具有较好的基因递药功能;作用4小时后的实验结果与1小时基本一致。因此,pag/nr-fam-sirna细胞内吞速度快于nr和fam-sirna本身,从而证明了本发明制备的pag共聚物具有较好的同时对药物和基因递药功能,有望开发为用于治疗肿瘤的药物和基因的共递送载体。11.2流式细胞术按照11.1描述的方法将培养的mcf-7细胞按2×105个/孔种于12孔板;细胞于37℃、5%co2培养箱中孵育24小时后,分别加入游离nr、pag/nr、pag/nr-mmp-9胶束孵育1和4小时;吸去上清液,用pbs洗三遍,0.25%胰酶消化,收集细胞,离心,去掉上清液,加入pbs制成细胞混悬液;以氩离子激光器作为光源,利用流式细胞仪对细胞进行分析,测定nr在细胞中的平均荧光强度,以x-mean值表示;具体结果如图11所示。图11为nr,pag-nr,mmp-9sirna,pag/nr-mmp-9在mcf-7细胞内吞实验流式结果图;具体的图11a为孵育1小时,nr,pag-nr,mmp-9sirna,pag/nr-mmp-9的荧光强度曲线;图11b为孵育4小时,nr,pag-nr,mmp-9sirna,pag/nr-mmp-9的荧光强度曲线;图11c为孵育1小时和4小时时nr,pag-nr,mmp-9sirna,pag/nr-mmp-9摄取量;从图11a可见,孵育1小时候,pag/nr和pag/nr-mmp-9的荧光强度分别是游离nr荧光强度的3.68和3.67倍;从图11c可见,mcf-7细胞对pag/nr和pag/nr-mmp-9的摄取量明显高于游离nr,综合图11可见,4小时后的实验结果与1小时基本一致;实验结果与利用激光共聚焦显微镜考察的结果相吻合;进一步说明pag共聚物具有较好的同时对药物和基因递药功能,有望开发为用于治疗肿瘤的药物和基因的共递送载体。实施例12:体内活体荧光成像实验及体内组织分布实验spf级balb/c裸鼠3只,右前肢腋下接种收集好的生长状态良好的人乳腺癌mcf-7细胞,预先将荧光材料icg代替dtx制备载药胶束pag/icg,待肿瘤体积长到约300mm3,将200微升样品溶液(生理盐水、icg、pag/icg)经尾静脉分别注射到小鼠体内;注射前和注射后0.5、2h、4h、8h和24h时间点麻醉状态下置于活体荧光成像仪中拍照成像;其中icg激发波长为780nm,发射滤光为800nm。曝光时间为30s;实验后的裸鼠断颈处死,取出心、肝、脾、肺、肾和肿瘤组织,pbs清洗后,滤纸吸干表面液体,活体荧光成像仪下进行拍照;具体实验结果如图12所示。图12为pag/icg、icg、空白裸鼠小动物成像实验结果;图12a是注射前和注射后0.5、2h、4h、8h和24h时间点麻醉状态下活体荧光成像;图12b是试验后裸鼠心、肝、脾、肺、肾和肿瘤组织处的荧光成像;图12c是空白、icg、pag/icg组肿瘤组织处的荧光成像。本实验所选用的icg荧光材料属于两亲性材料,几乎不受生物组织本底影响,不伤害实验动物本身,能够穿透深层组织产生信号进行成像,从图中可见,空白组裸鼠中几乎观测不到荧光干扰,icg、pag/icg经尾静脉进入裸鼠后,快速分布,且pag/icg给药组0.5小时后即可观察到icg荧光达到组织部位,同时24小时后取裸鼠心、肝、脾、肺、肾、肿瘤组织发现icg主要集中于肝和肿瘤组织,且其中pag/icg组肿瘤组织的荧光明显强于icg组。由此可见,本发明的pag具有较好的抗肿瘤作用。实施例13:pag载体药物对mcf-7荷瘤裸鼠的治疗作用实验24只裸鼠,雌性,体重18-20g左右,饲养于spf环境中,裸鼠食料、饮用水、垫料均定期经高压灭菌消毒,鼠笼、垫料一周更换一次;采用人源的mcf-7胰腺癌细胞建立裸鼠皮下移植瘤模型,将2*106的细胞悬浮于20ml新鲜的的pbs中,将细胞悬液注射至裸鼠右前肢腋下,10天后观察荷瘤裸鼠的种瘤体积达到60mm3,开始随机分为4组:①生理盐水模型组;②pag-mmp-9(14:1)治疗组;③pag/dtx-mmp-9(9:1:0.64)治疗组;④(多西他赛)注射液治疗组,每组6只;经尾静脉分别将pag/mmp-9、pag/dtx/mmp-9或注射到裸鼠体内,生理盐水模型组则直接尾静脉注射200μl生理盐水处理,给药剂量dtx为3mg/kg/次与mmp-9均为1.9mg/kg/次;将开始治疗的时间定义为第10天,隔天给药,至第40天;隔天观察各组裸鼠肿瘤大小、体重、进食情况和大小便情况,其中肿瘤大小用游标卡尺测量,分别记录每个肿瘤的长径(a)和短径(b),以公式v(mm3)=0.5*a2*b计算肿瘤大小;实验至40天终止,通过颈椎脱臼处死动物,取材。13.1病理检查切取裸鼠心、肝、脾、肺、肾、肿瘤组织,在4%中性多聚甲醛溶液(福尔马林)固定2天后,进行病理检查。60℃下浸蜡5h进行包埋处理,用石蜡切片机将组织蜡块切成4μm厚度的薄片,展片后在60℃温箱中放置2h;依次用浓度梯度乙醇浸泡脱水(70%乙醇、80%乙醇、90%乙醇),每级乙醇浸泡30分钟,之后用95%乙醇两次,每次20分钟,用苏木素染色8分钟,用自来水冲洗约8分钟,用1%盐酸酒精浸泡分化5s后,再用自来水冲洗8分钟,温热水蓝化后再使用自来水充分冲洗,最后进行伊红染色,封片后在光学显微镜下观察。13.2数据统计学分析实验数据以平均数±标准差(x±sd)进行表示,通过spss12.0统计学软件进行数据处理,组间比较采用单因素方差分析;p<0.05表示差异具有统计学意义。13.3实验结果图13为pag/dtx-mmp-9、pag-mmp-9、抗mcf-7乳腺癌的药效结果;其中13a为mcf-7荷瘤裸鼠肿瘤生长情况;13b为荷瘤裸鼠体重变化;13c为荷瘤裸鼠肿瘤重量比较;13d为荷瘤裸鼠肿瘤图片;与模型组比较,*p<0.05,**p<0.01;从图中可见,mcf-7荷瘤裸鼠经过1个月治疗后,pag/dtx-mmp-9组的抑瘤效果最为显著,其次为pag-mmp-9和组,抑瘤率分别为45.57%、37.8%和24.85%;瘤重数据结果表明,与生理盐水模型组比较,pag/dtx-mmp-9和pag-mmp-9均有显著性抑制作用;而且在治疗过程,未见裸鼠体重发生显著性变化;可见pag/dtx-mmp-9、pag-mmp-9对mcf-7乳腺癌具有较好的抑瘤作用,pag在整个治疗过程中,起到较好运载药物和基因的作用。对各组荷瘤裸鼠中肿瘤组织和脏器组织进行石蜡切片,苏木精-伊红(h&e)染色后组织病理学观察,结果如图14所示。图14为mcf-7荷瘤裸鼠心、肝、脾、肺、肾、肿瘤h&e组织切片图;从图中可见,生理盐水模型组肿瘤细胞密集,细胞核完整、包浆面积较小,肿瘤细胞没有发生凋亡和死亡;dtx组细胞中包浆面积增大,细胞核的密度相对于生理盐水组明显的减少,说明部分肿瘤细胞发生凋亡与坏死作用,而pag/dtx-mmp-9、pag-mmp-9组肿瘤组织细胞中包浆面积显著增大,细胞核显著减少,说明肿瘤细胞发生明显凋亡,肿瘤组织发生坏死;通过显微镜观察荷瘤裸鼠的心、肝、脾、肺、肾组织的h&e染色切片可得,各给药组与生理盐水模型对照组相比,没有发现明显的病理特征如内皮细胞的损伤、坏死、炎症反应以及再生性的改变等。总之,pag/dtx-mmp-9、pag-mmp-9对mcf-7乳腺癌具有较好的治疗作用,而且pag材料在治疗过程中可较好的运载药物和基因,显著性增强dtx对乳腺癌的治疗作用。综上所述:本发明直接由带羧基或与乙酸酐衍生化后带羧基的抗肿瘤活性药物x作为疏水端与亲水的超支化聚酰胺胺paas通过酰胺化反应得到氧化还原响应性两亲性共聚物pax,pax的主链上含有抗肿瘤活性药物,自身即具有抗肿瘤作用;pax可在水中自组装形成胶束,形成的胶束内层疏水端可进一步包裹另一种疏水性抗肿瘤药物y,并且由于胶束外层带有正电,通过静电作用,还可以进一步负载一种治疗基因z,得到负载治疗基因的胶束;总之pax及由pax自组装形成的胶束和负载治疗基因的胶束,都具有抗肿瘤作用,尤其是负载治疗基因的胶束可用作药物和基因的共递送载体,用于制备抗肿瘤药物,治疗包括肺癌、肝癌、乳腺癌、胰腺癌、肠癌等癌症;由pax得到的胶束pax/y/z,稳定性好,其结构中的双硫键具有氧化应激功能,可在肿瘤细胞中高浓度gsh的作用下发生解离,并释放出所载的药物x、y和基因z,从而实现靶向性,提高药物的生物利用度同时降低毒副作用;另外,本发明制备方法简单,无需特殊设备和苛刻条件,易于实现规模化生产,具有极强的实用价值,相对于现有技术而言,取得了显著性进步和出乎意料的效果。最后需要在此指出的是:以上仅是本发明的部分优选实施例,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容做出的一些非本质的改进和调整均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1