抗病毒药物组合物的制作方法
本发明涉及抗病毒药物领域,具体涉及抗病毒药物组合物。
背景技术:
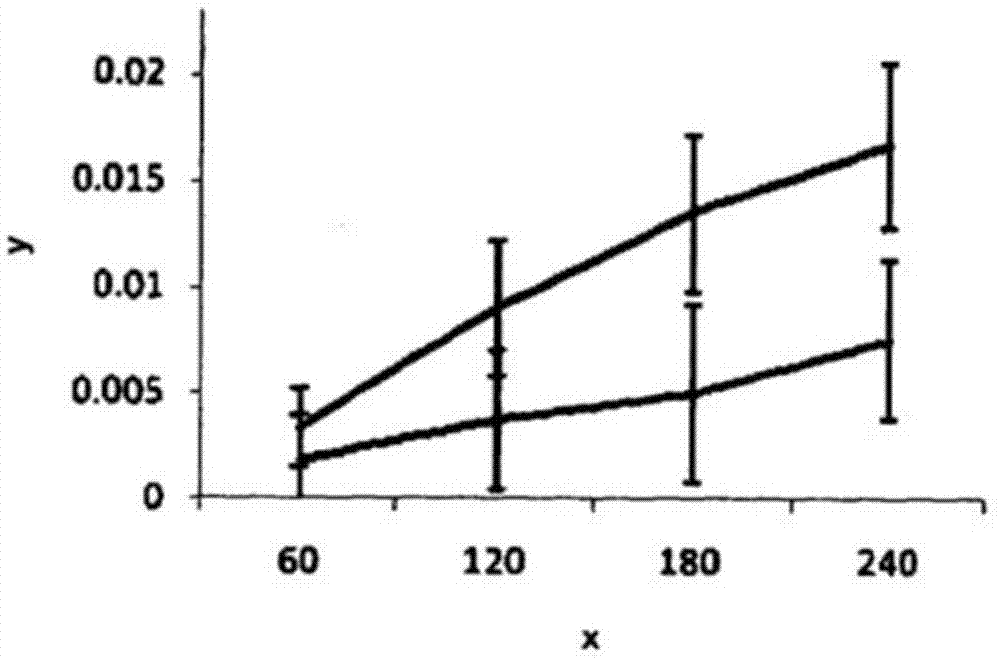
逆转录病毒属于病毒的逆转录病毒科。逆转录病毒是在宿主细胞中通过反转录酶被复制以从它的rna基因组产生脱氧核糖核酸(dna)的核糖核酸(rna)病毒。通常,dna会转录成rna,并且rna会翻译成蛋白质。然而,当宿主细胞被逆转录病毒感染时,转录过程相反,而且得到的dna随后通过整合酶插入宿主的基因组。该病毒此后复制作为宿主细胞的dna的部分。已知逆转录病毒导致某些类型的癌症,例如在人类和动物中均存在的白血病,以及一系列病毒感染,包括引起aids的病毒hiv。hiv和aids是普遍的疾病,在世界上每年引起很多人死亡。估计在2008年死于aids的人数为两百万,而同年活着携带hiv/aids的总人数为3340万。已有超过2500万人被认为死于hiv/aids,而且估计感染事件在全球仍将增加,包括在发达国家。现在认为异性恋传播已经超过同性恋传播以及认为母婴传播是感染的主要途径。同时已经研发药物用于治疗hiv和aids,如下面段落将描述的,存在普遍并长期存在的问题:药物的水溶性差使得它们的给药困难以及它们的生物利用度低。已经研发抗病毒药物和药剂并用于对抗病毒和逆转录病毒;这些药物已知分别为“抗病毒药物”和“抗逆转录病毒药物”。已知用于治疗hiv/aids的抗逆转录病毒的药物种类包括:-蛋白酶抑制剂(pis),通过抑制蛋白酶的活性靶向病毒装配,蛋白酶是参与hiv复制过程的酶;-核苷类逆转录酶抑制剂(nrtis),核苷酸逆转录酶抑制剂(ntrtis)和非核苷类逆转录酶抑制剂(nnrtis),所有这些抑制逆转录酶的活性;-整合酶抑制剂,设计用于阻断整合酶的活动,整合酶是将病毒基因组插入到宿主细胞的dna中的病毒酶;-进入抑制剂,也称为融合抑制剂,干涉hiv结合、融合和进入到宿主细胞;-成熟抑制剂,也抑制蛋白酶的活性。这些抗病毒和抗逆转录病毒药物通常以组合的方式给出并且因此技术给提高患者依从性并可能降低毒性或药物和药物之间的相互作用提供了机会,该技术能够提高活性、生物利用度和配方范围(以产生组合药物形式)。hiv和aids药物既可以用于疾病的预防也可以用于疾病的治疗。aids晚期的患者可能不能吞咽药片。因此想要获得以液体形式可用的抗逆转录病毒药物以提供可注射的药剂或允许直肠给药。液体形式的药剂也允许剂量不断的变化,这对儿童的治疗特别有益,因为剂量可能根据体重计算。已经有很多现有技术尝试提高不同抗病毒药物的水溶性,包括用于治疗hiv/aids的抗逆转录病毒药物。wo03/084462描述了一种通过加入胡椒碱制备具有改进的生物利用度的抗逆转录病毒蛋白酶抑制剂的方法。该加入胡椒碱的药物组合物可以以液体、粉末、胶囊、片剂形式或其他任何想要的剂形制备。不幸的是这种方法不适用于所有的蛋白酶抑制剂,例如,它不适用于利托那韦,因为它不是通用的因此受限。ep0872233描述了洛韦胺的药物组合物,其中剂形具有高的药物含量。这些组合物包括熔融挤出包括洛韦胺和适量的水溶性聚合物的混合物以及随后碾碎所述熔融挤出的混合物而获得的颗粒。不幸的是描述的熔融挤出的方法也不通用,因为包括在表现出任意程度的热敏感性的最终组合物中的这些抗逆转录病毒药物或其他在药学上可接受的赋形剂不适用于这种方法。类似地,wo2008/017867描述了使用熔融挤出方法制备的抗逆转录病毒固体口服组合物的规定。现有技术说明确实需要提供另外的不溶于水的抗逆转录病毒药物和更普遍的抗病毒药物,以可溶的形式,并且以这种方式提供生理上可接受的溶液。此外可能想要提供抗病毒药物和抗逆转录病毒药物,这些药物相对于它们现在可获得的形式表现出提高的生物利用度。特别种类的不溶于水的问题不限于抗病毒和抗逆转录病毒药物,并且甚至在其他领域,已经提出很多技术以规划另外的不溶于水的种类。例如,wo2004/011537描述了固体的形成,多孔珠子包括三维开室晶格的水溶性聚合材料。这些是典型的“乳液模板”材料通过从高内相比乳液(highinternalphaseemulsion)(hipe)除去水(连续的,水相)和非水分散(或不连续)相形成,该“乳液模板”材料具有水溶性聚合物溶在连续水相中。珠子通过滴加hipe乳液到低温流体(例如液氮),然后通过冷冻干燥形成在低温流体中的颗粒以去除大量的连续水相和非水分散相,剩下骨架结构形式的聚合物形成。珠子在水中快速溶解。有利地,溶解在乳液的非水相的不溶于水的组分在它冷冻和干燥之前分散在溶解珠子的聚合物骨架的水中。在优先权日,以及也在提交日,不能想象在wo2004/011537中描述的方法可以适用于抗病毒药物(包括抗逆转录病毒药物)的应用,并且因此没有提到或表明本发明对抗病毒药物的应用。wo2005/011636公开了非乳液基的喷雾干燥方法用于在聚合物中形成药物的‘固体无定形分散体’。在这种方法中,将聚合物和低溶解度的药物溶解在溶剂中并喷雾干燥以形成分散体,其中,药物大部分以无定形形式而不是以晶体形式存在。wo2006/079409和wo2008/006712各自描述了怎样制备不溶于水的材料在水中形成纳米分散体,优选通过喷雾干燥的方法。在wo2006/079409中,将不溶于水的材料溶解在乳液的溶剂相(即“油”相)中,同时将水溶性结构化试剂溶解在乳液的水相中。在wo2008/006712中,将不溶于水的材料溶解在单相混合的溶剂体系中并作为水溶性结构化试剂在相同相共存。在两种情况中将液体(即,乳液或单相混合的溶剂)在高于环境温度下干燥,例如通过喷雾干燥以制备结构化试剂的粉末颗粒,作为载体,具有不溶于水的材料分散其中。当将这些粉末颗粒置于水中时,水溶性结构化试剂溶解以形成不溶于水的材料的纳米分散体,具有在水中具有通常小于300nm的z-均粒径的所述纳米颗粒。不溶于水的材料表现好像它在溶液中。wo2006/079409显示通过公开的方法制备的荧光剂材料比那些通过已知的冷冻干燥方法制备的表现出更好的性能。wo2008/006712明确triclosantm纳米分散体具有额外的益处,就重量而言(weightforweight),即使在非常低的浓度下,triclosantm纳米分散体比通常期望的triclosantm更有效。技术实现要素:本发明涉及含有一种或多种抗病毒药物组合物。具体地,本发明涉及含有用于治疗由病毒引起的感染的药物(即抗病毒药物)的药学上有效的组合物(和它的前体)。本发明特别(但不是特定)涉及含有用于治疗由逆转录病毒引起的感染的药物(即抗逆转录病毒药物)的组合物。本发明进一步涉及药学上可接受的形式的抗病毒药物,不管是单独还是与另一种药物(例如另一种抗病毒药物或其他药学上可接受的赋形剂)组合。通常认为本发明可应用的组合物含有任意抗病毒药物(即,用于治疗由病毒,例如疱疹病毒、肝炎b、肝炎c、流感等引起的感染),尤其是那些含有抗逆转录病毒药物(即,用于治疗由逆转录病毒引起的感染)的组合物,但是将特别描述关于含有用于治疗人类免疫缺陷病毒(hiv)和获得性免疫缺陷综合症(aids)的药物的组合物。然而,在任意的这些现有出版物中没有公开或表明本文公开的技术可能或可以应用于抗病毒药物以有效的“溶解”它们。出人意料地,因此,本发明的发明人现在已经确定乳液基(emulsion-based)方法和单相方法都用于生产能够在水中形成不溶于水的材料的纳米分散体的颗粒,如上讨论的,能够用于生产水分散形式的一种或多种抗病毒药物,包括抗逆转录病毒药物,特别是用于治疗hiv和aids的相对不溶于水的抗逆转录病毒药物。附图说明图1示出了溶解在dmso中的沙奎那韦,相对时间(在x轴以分钟计)转移小室中顶端侧(下面的线)相对于基底侧(上面的线)的药物浓度(在y轴以微摩尔计)。图2-图6示出了以纳米分散形式制备的沙奎那韦,相对时间转移小室中顶端侧(下面的线)相对于基底侧(上面的线)的药物浓度(在y轴以微摩尔计)。图7示出了从顶端侧到基底侧的表观渗透率(pappa-b)相对于从基底侧到顶端侧的表观渗透率(pappb-a)的比率。具体实施方式第一方面因此,本发明的第一方面提供一种组合物,该组合物包括至少一种不溶于水的抗病毒药物和至少一种水溶性的载体材料,其中,所述不溶于水的抗病毒药物通过所述水溶性的载体材料以纳米分散形式分散。本发明的第一方面尤其适用于不溶于水的抗逆转录病毒药物。第二方面本发明的第二方面提供一种抗病毒药物制剂,该抗病毒药物制剂包括至少一种不溶于水的抗病毒药物和至少一种水溶性的载体材料,其中,所述不溶于水的抗病毒药物通过所述水溶性的载体材料以纳米分散形式分散。本发明的第二方面也尤其适用于不溶于水的抗逆转录病毒药物。粒径贯穿说明书,通过“纳米分散”和类似术语我们定义分散体,其中,z-均粒径(直径)(不然称为水力学直径)小于1000nm。优选地,不溶于水的抗病毒药物的纳米分散形式的z-均直径优选为800nm以下,甚至更优选为500nm以下,尤其200nm以下,以及最优选100nm以下。例如,不溶于水的抗病毒药物的纳米分散形式的z-均直径可以在50-750nm范围内。测量本发明的分散产品的粒径的优选方法使用动态光散射(dynamiclightscattering)(dls)仪器(纳米s,由英国马尔文仪器制造(nanos,manufacturedbymalverninstrumentsuk))。具体地,马尔文仪器纳米s使用红外(633nm)4mw氦氖激光照亮含有将被测量的悬浮的颗粒的标准光学质量紫外(uv)试管。本申请中引用的粒径是使用仪器制造商提供的标准科学实验报告用设备获得的。纳米颗粒在干燥固体材料中的粒径,例如不溶于水的抗病毒药物纳米颗粒的粒径,是从测量干燥固体材料被分散在水中得到的粒径推理出来的。在纳米分散体中的不溶于水的抗病毒药物的粒径被认为带来很多重要的伴随的益处,包括具有更小粒径的纳米分散体被认为比具有较大粒径的纳米分散体更稳定,那样,当载体材料在水性介质中溶解时,不溶于水的药物能够发生非常快的分散,优选在5分钟内被引入水性介质中。颗粒的尺寸是指在某些情况下能够达到清水分散,即在水性介质中分散的药物颗粒肉眼看不见而且液体呈现清澈。此外,很多药物代谢动力学优势与本发明的产品的纳米分散规模相关。认为根据本发明的组合物和抗病毒药物制剂都具有增加的生物利用度(即,与药物以已知形式给药相比,增加量的药物使得药物以吞咽量的比例进入受试者的血流)表明如下:-auc增加,即,在24小时周期中药物总的暴露量更大;-一次给药后的最大血药浓度(c-max)增加,即,药物在血液中的最大浓度更高;-c-max更早,即,药物在血流中达到最大浓度所花费的时间缩短;-药物的半衰期增加,即,药物的血液浓度降至它的c-max的50%所花费的时长;以及-禁食的(fasted)和非禁食的(non-fasted)受试者之间的变异性降低。抗病毒药物如上陈述,包括在本发明的组合物和药物制剂中的抗病毒药物是不溶于水的抗病毒药物。在本发明的上下文中,应用于抗病毒药物的“不溶于水的”是指在环境温度和压力下药物在水中的溶解度小于10g/l。作为比较,根据吉利德科学公司(gileadsciencesinc.)的m.法尔迪(m.fardis)和r.欧利耶(r.oliyai)在他们题为“5.20:富马酸替诺福韦(tenofovirdisoproxilfumarate):替诺福韦的口服前药(anoralprodrugoftenofovir)”;的论文652页中指出的,替诺福韦的口服前药,富马酸替诺福韦,具有13.4mg/ml(即,13.4g/l)的高的水溶解度。为了避免任何怀疑,在本申请中术语“环境温度”是指25℃同时“环境压力”是指1个大气压(101.325kpa)。优选地,不溶于水的抗病毒药物在环境温度和压力下在水中具有小于5g/l的溶解度,更优选小于1g/l,甚至更优选小于150mg/l,以及特别优选小于100mg/l。这种溶解度水平提供了什么是本说明书中的“不溶于水的”的预期解释。优选的不溶于水的抗病毒药物包括治疗由疱疹病毒、肝炎病毒、流感病毒和逆转录病毒引起的感染的药物。优选的不溶于水的抗逆转录病毒药物包括,但不限于,蛋白酶类抑制剂(pis)、核苷类逆转录酶抑制剂(nrtis)、核苷酸类逆转录酶抑制剂(ntrtis)、非核苷类逆转录酶抑制剂(nnrtis)、整合酶抑制剂、进入抑制剂、成熟抑制剂和它们在药学上可接受的盐和前体。不溶于水的抗病毒药物可以有利地选自下述中的一种或多种:-阿昔洛韦(aciclovir)、二十二醇(docosanol)、依度尿苷(edoxudine)、泛昔洛韦(famciclovir)、膦甲酸(foscarnet)、疱疹净(idoxuridine)、喷昔洛韦(penciclovir)、三氟尿苷(trifluridine)、容满它定(tromantidine)、缬昔洛韦(valaciclovir)和阿糖腺苷(vidarabine)(所有这些治疗由一种或多种疱疹病毒引起的感染);-阿德福韦(adefovir)、波西普韦(boceprevir)、恩替卡韦(entecavir)、利巴韦林(ribavirin)和塔利韦林(taribavirín)(所有这些治疗由一种或多种肝炎病毒引起的感染);-三环癸胺(amantadine)、阿比朵尔(arbídol)、奥塞米韦(oseltamívir)、帕拉米韦(peramívír)、金刚乙胺(rimantidine)和扎那米韦(zanamívír)(所有这些治疗由一种或多种流感病毒引起的感染)。不溶于水的抗逆转录病毒药物可以有利地选自下述中的一种或多种:-安瑞那韦(amprenavir)、阿扎那韦(atazanavir)、地瑞那韦(darunavir)、福沙那韦(fosamprenavir)、印地那韦(indinavir)、洛匹那韦(lopinavir)、那非那韦(nelfinavir)、利托那韦(ritonavir)、沙奎那韦(saquinavir)和替拉那韦(típranavir)(所有这些都是蛋白酶抑制剂);-阿巴卡韦(abacavir)(abc)、安都修韦(amdoxovir)(amdoxovir)、阿立他滨(apricitabine)(atc)、地丹诺辛(didanosine)(ddi)、艾夫他滨(elvucitabíne)、恩曲他滨(emtricitabine)(ftc)、恩替卡韦(inn)、拉米夫定(lamivudine)(3tc)、冉次韦(racivir)(racivir)、斯坦匹定(stampidine)、司他夫定(stavudine)(d4t)、扎西他滨(zalcitabine)(ddc)和齐多呋定(zidovudine)(azt)(所有这些都是nrtis);-阿德福韦(也称为双-pompmpa)和替诺福韦(tenofovir)(两者都是ntrtis);-地拉夫定(delavirdine)、依法韦仑(efavirenz)、依曲韦林(etravirine)、赖斯韦林(lersivirine)、洛韦胺(loviride)、奈韦拉平(nevirapine)和日匹韦林(rilpivirine)(所有这些都是nnrtis);-埃替格韦(elvitegravir)、木脂素(globoidnana)、gsk-572、mk-2048和雷特格韦(raltegravir)(所有这些都是整合酶抑制剂);-恩伏韦日体得(enfuviritide)、巴立组麻巴(ibalizumab)、马拉维若(maraviroc)和韦日维若(vicriviroc)(所有这些都是融合/进入抑制剂);-贝韦立马(bevirimat)和韦未从(vivecon)(两者都是成熟抑制剂);和它们在药学上可接受的盐和前体以及它们的混合物。可以使用任意的合适的抗病毒药物的在药学上可接受的盐,该盐可以是本领域技术人员已知的盐。类似地,可以使用任意的合适的抗病毒药物的前体,该前体可以是本领域技术人员已知的前体。例如,合适的前体可以是前体药物形式,我们是指在受试者中分解释放出活性抗病毒药物的化合物。根据本发明的组合物和药物制剂可以含有两种或多种抗病毒药物、它们在药学上可接受的盐和/或前体的混合物。任意这种附加的抗病毒药物可以是水溶性的(例如,恩曲他滨,是nrti)并且可以随着水溶性的载体材料引入组合物或药物制剂。此外,它可以具有表面活性剂的性质。在逆转录病毒感染的环境中,使用这种药物的混合物通常参考高活性抗逆转录病毒疗法(haart)。因为抗逆转录病毒药物的组合被认为对hiv复制产生多重障碍并且改善治疗,因此haart的使用是典型的hiv和aids的治疗。此外,使用haart,可以抑制一种或多种药物的抗药性的发展。本发明的组合物和药物制剂优选为基本上无溶剂。在本发明的上下文中,术语“基本上无溶剂”是指组合物和药物制剂的自由溶剂含量小于15%,优选10%以下,更优选5%以下以及最优选2%以下。为了避免怀疑,贯穿整篇说明书,所有的百分比是重量百分比,除非另有说明。载体材料如上陈述,包括在本发明的组合物和药物制剂中的载体材料为水溶性载体材料。在本发明的上下文中,用于载体材料的“水溶性”是指载体材料在环境温度和压力下在水中的溶解度至少为10g/l。术语“水溶性”包括结构水相以及分子的单分散种类的真离子溶液的形成。出于比较,根据j.pharma.sci.,2001,90(8);1015-25,利托那韦水溶性较差,在ph6.8和37℃具有1μg/ml(即,0.001g/l)的溶解度。如在此所述,优选的水溶性载体材料可以选自水溶性无机材料、水溶性表面活性剂、水溶性聚合物、水溶性糖和它们的混合物中的一种或多种。聚合物载体材料合适的水溶性聚合物载体材料的例子包括:(a)天然聚合物(例如自然产生的橡胶如瓜尔胶、藻酸盐、刺槐豆胶或多糖(例如右旋糖酐));(b)纤维素衍生物,例如黄原胶、木葡聚糖、醋酸纤维素、甲基纤维素、羟乙基纤维素、羟乙基甲基纤维素、羟丙纤维素(hpc)、羟丙基甲基纤维素(hpmc)、羧甲基纤维素和它的盐(例如钠盐-scmc),或羧甲基羟乙基纤维素和他的盐(例如钠盐);(c)从两种或多种单体制备的均聚物或共聚物选自:乙烯醇、丙烯酸、甲基丙烯酸、丙烯酰胺、甲基丙烯酰胺、丙烯酰胺甲基丙烷磺酸酯、氨基烷基丙烯酸酯、氨基烷基-甲基丙烯酸酯、丙烯酸羟乙酯、甲基丙烯酸羟乙酯、乙烯基吡咯烷酮、乙烯基咪唑、乙烯基胺、乙二醇和其他烯化醇、乙烯氧化物和其他烯化氧化物、乙撑亚胺、苯乙烯磺酸酯、丙烯酸乙二醇酯和甲基丙烯酸乙二醇酯;(d)环式糊精,例如β-环式糊精;(e)它们的混合物。为了避免任何怀疑,已知上述确定的种类中的一些具有水溶性和不溶于水的两种形式,例如,醋酸纤维素,它的溶解度根据它的乙酰基团取代(ds)度变化。然而,为了本发明的目的,应该理解成仅考虑任意这些种类的水溶性形式。当聚合的材料是共聚物时,它可能是统计共聚物(在此之前也称为随机共聚物)、嵌段共聚物、接枝共聚物或超支化共聚物。如果它们的存在不破坏得到的聚合的材料的水溶性或水分散性,除了那些列出的,也可以包括非上述那些列出的共单体。合适的和优选的均聚物的例子包括聚乙烯醇(pva)、聚丙烯酸、聚甲基丙烯酸、聚丙烯酰胺(例如聚-n-异丙基丙烯酰胺)、聚甲基丙烯酰胺、聚丙烯酰基胺(polyacrylamines)、聚甲基丙烯酰基胺(polymethylacrylamines)(例如聚甲基丙烯酸二甲基氨基乙酯和聚-n-吗啉代甲基丙烯酸乙酯)、聚乙烯吡咯烷酮(pvp)、聚苯乙烯磺酸酯、聚乙烯基咪唑、聚乙烯基吡啶、聚-2-乙基噁唑啉、聚乙烯亚胺和它们的乙氧基衍生物。一方面,聚乙二醇(peg)、聚乙烯吡咯烷酮(pvp)、聚(2-乙基-2-噁唑啉)、聚乙烯醇(pva)、羟丙纤维素(hpc)和羟丙基甲基纤维素(hpmc)和藻酸盐为优选的聚合物载体材料。另一方面,聚乙烯吡咯烷酮(pvp)、聚乙烯醇(pva)、羟丙纤维素(hpc)和羟丙基甲基纤维素(hpmc)为优选的聚合物载体材料。为了避免任何怀疑,如果聚合物载体材料用于本发明,它基本上没有,而且优选完全没有交联,因为载体材料的目的是与水介质接触溶解。众所周知交联对聚合物的物理性质具有大的影响,因为交联限制了聚合物链的相对移动性,增加了分子量并引起大范围的网形成,因此阻碍了聚合物的溶解能力。例如,聚苯乙烯溶解在很多溶剂例如苯、甲苯和四氯化碳中。然而即使用少量的交联剂(二乙烯基苯,0.1%),聚合物仅仅膨胀不再溶解。表面活性剂载体材料载体材料为表面活性剂,该表面活性剂可以为非离子的、阴离子的、阳离子的、两性的或两性离子的表面活性剂。合适的非离子的表面活性剂的例子包括乙氧基甘油三酸酯;脂肪醇乙氧基化物;烷基酚乙氧基化物;脂肪酸乙氧基化物;脂肪酰胺乙氧基化物;脂肪胺乙氧基化物;山梨聚糖链烷酸酯;乙基化山梨聚糖链烷酸酯;烷基乙氧基化物;氧化乙烯和氧化丙烯的嵌段共聚物,即,泊洛沙姆(以商品名pluronicstm使用);烷基多糖甙(alkylpolyglucosides);油脂剂乙氧基化物;烷基多糖苷(alkylpolyglycosides)。合适的阴离子的表面活性剂的例子包括烷基醚磺酸盐;烷基醚羧酸盐;烷基苯磺酸盐;烷基醚磷酸盐;二烷基磺基琥铂酸盐;肌氨酸盐;烷基磺酸盐;皂;烷基硫酸盐;烷基羧酸盐;烷基磷酸盐;石蜡磺酸盐;仲正烷基磺酸盐(secondaryn-alkanesulfonates);α-烯烃磺酸盐;羟乙基磺酸盐磺酸盐(isethionatesulfonates)。合适的阳离子的表面活性剂的例子包括脂肪胺盐;脂肪二胺盐;季胺化合物;膦表面活性剂;锍表面活性剂。合适的两性离子的表面活性剂的例子包括氨基酸的n-烷基衍生物(例如甘氨酸、甜菜碱、丙氨酸);咪唑啉表面活性剂;氧化胺;氨基甜菜碱。可以使用表面活性剂的混合物;在这些混合物中个别组分可以是液体。特别优选烷氧基化非离子表面活性剂(尤其是泊洛沙姆,例如pluronictm型材料)、烷基磺酸盐、烷基硫酸盐(尤其是十二烷基硫酸钠(sds))、脱氧胆酸钠、十四(烷)酸钠、多库酯钠、酯表面活性剂(优选非-peg-化的(non-peg-ylated)山梨聚糖的酯(所谓的spantm酯)以及聚山梨醇酯(是peg-化山梨聚糖的酯)(所谓的tweentm酯))以及阳离子的表面活性剂(尤其是十六烷基三甲基溴化铵-ctab)作为表面活性剂载体材料。无机载体材料载体材料也可以为既不是表面活性剂也不是聚合物的水溶性的无机材料。已经发现简单的有机盐适合,特别是与上述描述的聚合的和/或表面活性剂载体材料混合。合适的盐包括碳酸盐、重碳酸盐、卤化物、硫酸盐、硝酸盐和醋酸盐,特别是钠、钾和镁的可溶性盐。优选的材料包括碳酸钠、重碳酸钠和硫酸钠。这些材料具有廉价和生理上可接受的优势。它们也是相对惰性的同时与在制药产品中发现的很多材料可以相容。有机载体材料载体材料也可以为既不是表面活性剂也不是聚合物也不是无机载体材料的水溶性的小有机材料。已经发现简单的有机糖适合,特别是与上述描述的聚合的和/或表面活性剂载体材料混合。合适的小有机材料包括甘露醇、木糖醇和菊粉,等等。根据本发明的组合物和药物制剂可以包括两种或多种载体材料。载体材料的混合物是有优势的。优选的混合物包括表面活性剂和聚合物的组合,例如混合物优选包括至少一种:a)聚乙烯醇(pva)、聚乙二醇(peg)、聚乙烯吡咯烷酮(pvp)、羟丙基纤维素和羟丙基甲基纤维素(hpmc)、藻酸盐和至少一种;b)烷氧基化的非离子的表面活性剂(尤其是泊洛沙姆,例如pluronictm型材料)、烷基磺酸盐、烷基硫酸盐(尤其是十二烷基硫酸钠(sds))、脱氧胆酸钠、十四(烷)酸钠、多库酯钠、酯表面活性剂(优选非-peg-化的山梨聚糖的酯(所谓的spantm酯)以及聚山梨醇酯(是peg-化的山梨聚糖的酯(所谓的tweentm酯))以及阳离子的表面活性剂(尤其是十六烷基三甲基溴化铵-ctab)。第三方面本发明的第三方面提供至少一种不溶于水的抗病毒药物和至少一种水溶性载体材料的水性纳米分散体,所述纳米分散体通过水和本文所述的组合物或药物制剂结合而获得。本发明的组合物或药物制剂与水性介质(例如水)一混合,水溶性载体就溶解并且不溶于水的抗病毒药物就分散,作为纳米分散体,以充分好的形式贯穿水性介质,在很多方面表现的像可溶的材料一样。本发明的第三方面尤其适用于不溶于水的抗逆转录病毒药物。不溶于水的材料在干燥产品中的粒径优选使得在水中分散的不溶于水的材料具有小于1000nm的z-均粒径,由本文描述的马尔文方法测定。以干燥固体粉末形式在水性介质中分散的抗病毒药物的纳米粒径被认为没有明显的减小。优选地,不溶于水的抗病毒药物的纳米分散形式的z-均直径小于1000nm,优选800nm以下,甚至更优选500nm以下,特别优选200nm以下,以及最特别优选100nm以下。例如,不溶于水的抗病毒药物的纳米分散形式的z-均直径可以在50-750nm范围内。下文提供的例子中已经测量的z-均粒径低至74nm。水性纳米分散体可以通过本领域技术人员认为的适合结合根据本发明的组合物或药物制剂与水性介质的已知的任意方法制备。关于上文提到的纳米分散体,优选的不溶于水的抗病毒药物和优选的水溶性载体材料是如上文描述的并且在下文做进一步的详细说明。纳米分散体的类似的优选的物理特性如上文所述。通过应用本发明的显著水平的‘不溶于水的’材料可以在很大程度上达到相当于真溶液的状态。当需要液体形式制药时,干燥的产品可以溶解在水性介质中以形成包括高达5%(以及优选大于0.1%,更优选大于0.5%和更优选大于1%)的不溶于水的材料的纳米分散体。当然本领域技术人员将领会在分散体中的不溶于水的材料的实际量将最终依赖于分散体给药的方式,例如,以注射形式、作为口服液体、以适合静脉注射给药的形式、直肠给药、通过鼻内喷雾,等等。展望水性纳米分散体将成为适合向受试者给药的形式,不论‘原本’或下面进一步稀释。可选地,水性分散体可以与其他活性材料结合以生产适合在结合治疗例如haart中使用的药物。第四方面本发明的第四方面提供制备包括至少一种不溶于水的抗病毒药物和至少一种水溶性的载体材料的组合物的方法,其中,不溶于水的抗病毒药物通过水溶性的载体材料以纳米分散形式分散,该方法包括如下步骤:(a)形成乳液,该乳液包括:(i)不溶于水的抗病毒药物在同样与水不混溶的溶剂中形成的溶液,以及(ii)水溶性的载体材料在水性溶剂中形成的溶液,以及(b)干燥乳液以去除水性溶剂和与水不混溶的溶剂以获得基本上无溶剂的抗病毒药物在载体材料中的纳米分散体。为了方便,这种类型的方法在此被称为“乳液”方法。在这种方法中,不溶于水的药物可以溶解在与水不混溶的,油的/有机溶剂中以形成油相,同时水溶性载体材料可以溶解在水性溶剂中以形成水相。然后可以通过结合油相和水相形成乳液,以已知的方式来形成水包油型(o/w)乳液,其中,油相为内部的/分散的/不连续的相以及水相为外部的/连续的相。本发明的第四方面尤其适用于不溶于水的抗逆转录病毒药物。优选地,非水(油)相包括从大约10%到大约95%体积比的乳液,更优选从大约20%到大约68%体积比。水性溶剂可以为水。乳液通常在本领域技术人员已知的条件下制备,例如,通过使用磁性搅拌棒、均质器,或超声(波)发生器。乳液不需要特别稳定,它们在干燥之前不进行广泛的相分离。在根据本发明的优选方法中,制备的水连续的乳液具有在500nm和5000nm之间的平均分散相微滴尺寸(使用马尔文峰强度)。超声处理也是降低乳液体系微滴尺寸的特别优选的方式。我们发现热体系超声(波)发生器xl在10级操作两分钟是合适的。第五方面本发明的第五方面提供制备包括至少一种不溶于水的抗病毒药物和至少一种水溶性的载体材料的组合物的方法,其中,不溶于水的抗病毒药物通过水溶性的载体材料以纳米分散形式分散,该方法包括如下步骤:(a)提供溶液,该溶液包括:(i)至少一种非水性溶剂,(ii)任选地,水性溶剂,(iii)水溶性的载体材料,该水溶性的载体材料在(i)和(ii)的混合物中是可溶的,以及(iv)不溶于水的抗病毒药物,该不溶于水的抗病毒药物在(i)和(ii)的混合物中是可溶的,而在单独的(ii)中是不溶的,以及(b)干燥溶液以去除水性溶剂(当存在时)和非水性溶剂以获得基本上无溶剂的抗病毒药物在载体材料中的纳米分散体。为了方便,这种类型的方法在此被称为“单相”方法。优选地,创造的混合物是单相溶液。在这种方法中,不溶于水的药物和水溶性载体材料都可以溶解在非水性溶剂(加上水性溶剂)(的混合物)中以形成单相溶液。本发明的第五方面尤其适用于不溶于水的抗逆转录病毒药物。在根据本发明的“单相”方法中,载体材料和不溶于水的抗病毒药物在非水性溶剂或这种溶剂与水性溶剂的混合物中都是可溶的。在这里和在说明书的别处,非水性溶剂可以为非水性溶剂的混合物。水性溶剂可以为水。在这种情况下,干燥步骤的原料可以为水溶性载体材料和不溶于水的抗病毒药物都溶解其中的单相材料。这种原料也可能为载体和药剂都溶解在相同相中的乳液。非水性溶剂在制备本发明的组合物中,使用非水性溶剂,即,易挥发的、非水性溶剂。这可能或者是在干燥之前的预混合中与其他溶剂(任选包括水性溶剂)混溶的非水性溶剂,或者是与水不能混溶的以及与水性溶剂一起可以形成乳液的非水性溶剂。在本发明的一种可选形式中,使用能够在抗病毒药物以及载体材料存在的情况下与水形成单相的单独的非水性溶剂。用于这些实施方式的优选的溶剂为极性、质子或非质子溶剂。特别优选的非水性溶剂选自卤仿(优选二氯甲烷,氯仿),低级(c1-c10)醇(优选甲醇、乙醇、异丙醇、异丁醇),有机酸(优选甲酸、乙酸),酰胺(优选甲酰胺、n,n-二甲基甲酰胺),腈(优选乙腈),酯(优选乙酸乙酯)、醛和酮(优选丁酮、丙酮),和与水混溶的种类包括杂原子(优选四氢呋喃、二烷基亚砜)。当然本领域技术人员将领会到这些之外,仅仅与水不能混溶的溶剂对于“乳液方法”是适合的并且这些可以适当的选择。卤仿、低级醇、酮和二烷基亚砜是最优选的非水性溶剂。在本发明的另一种可选形式中,非水性溶剂与水性溶剂不混溶并且形成乳液。乳液的非水性相优选选自下面易挥发的有机溶剂的组中的一种或多种:-烷类,优选庚烷、正己烷、异辛烷、十二烷、癸烷;-环烃,优选甲苯、二甲苯、环己烷;-卤化烷类,优选二氯甲烷、二氯乙烷、三氯甲烷(氯仿)、三氯氟甲烷和四氯乙烷;-酯,优选乙酸乙酯;-酮,优选2-丁酮;-醚,优选二乙醚;-易挥发的环硅酮,优选含有4到6个硅单元的或者线性或者环二甲基硅氧烷。合适的例子包括dc245和dc345,两者都获自道康宁公司(dowcorninginc)。优选的溶剂包括甲苯、环己烷和乙酸乙酯。优选的非水性溶剂,不论是否易混溶,具有小于150℃的沸点,以及更优选地,具有小于100℃的沸点,以便促进干燥,尤其是在实际条件下并且没有使用特别的设备的喷雾干燥。优选它们是不易燃的,或具有高于本发明的方法中所使用的温度的闪点。优选地,非水性溶剂包括从大约10%到大约95%体积比的任意乳液构成,更优选从大约20%到大约80%体积比。在单相方法中溶剂的量优选为20-100%体积比。特别优选的溶剂为醇,特别是乙醇和卤化溶剂,更优选含氯溶剂,最优选选自(二-或三-氯甲烷)的溶剂。助表面活性剂除了非水性溶剂之外,在干燥步骤之前,在组合物中可以使用任意的助表面活性剂。本发明的发明人已经确定添加相对少量的易挥发的助表面活性剂降低生产的纳米分散的抗病毒药物的颗粒直径。这对颗粒体积能够具有显著的影响。例如,从297nm降到252nm,相当于粒径降低了大约40%。因此,添加少量的助表面活性剂对于不改变最终产品配方而降低根据本发明的材料的粒径提供了简单的和便宜的方法。优选的助表面活性剂为沸点小于220℃的短链醇或胺。优选的助表面活性剂为线性醇。优选的助表面活性剂为伯醇和伯胺。特别优选的助表面活性剂选自3-6个碳的醇组成的组。合适的醇助表面活性剂包括正丙醇、正丁醇、正戊醇、正己醇、己胺和它们的混合物。优选地,助表面活性剂的存在量(以体积计)小于溶剂,优选地,溶剂和助表面活性剂之间的体积比在100:40到100:2的范围内,更优选100:30到100:5。在本发明的上下文中,必要的是载体材料和抗病毒药物在干燥步骤之前都是基本上充分溶解在它们各自的溶剂中,并且优选完全溶解。在本发明说明书的范围内没有教导浆体的干燥。为了避免任何怀疑,因此情况是在干燥步骤之前,乳液或混合物的固体含量使得为溶液中存在的可溶的材料的90%以上,优选95%以上,更优选98%以上,并且理想的是为100%。干燥优选地,干燥步骤同时去除水和其他溶剂。对于干燥中间乳液或溶液的最优选的方法为喷雾干燥。这在去除水性和非水性易挥发组分以将载体和抗病毒药物形成粉末形式特别有效。然而可选地,干燥可以通过冷冻干燥实现,冷冻干燥带来它自己特殊的益处,例如在制备用于静脉注射给药的无菌制剂中。喷雾干燥和冷冻干燥为本领域技术人员已知的技术。对于喷雾干燥,我们发现获自布琪(buchi)的b-290微型喷雾干燥器适合用于实验室喷雾干燥。对于大规模的喷雾干燥,获自geaniro的pharmasdtm喷雾干燥器是适合的。优选干燥温度应该在或高于100℃,优选高于120℃和最优选高于140℃。发现升高的干燥温度在再溶解的纳米分散材料中产生较小的颗粒。对于冷冻干燥,我们发现virtis台式bt4kzl冷冻干燥仪器适合用于实验室冷冻干燥,同时,获自生物制药工艺系统有限公司(biopharmaprocesssystemsltd)的usifroid冷冻干燥器适合用于大规模的冷冻干燥。干燥原料用于喷雾干燥或冷冻干燥的典型原料包括:a)表面活性剂,b)非水性溶剂,c)溶解在原料中的超过0.1%的至少一种不溶于水的抗病毒药物,d)聚合物,以及e)可选的水。因此,在本发明中,优选的原料包括:a)表面活性剂,选自:非离子泊洛沙姆(尤其是pluronictm材料)、烷基磺酸盐、烷基硫酸盐(尤其是sds)、脱氧胆酸钠、十四(烷)酸钠、多库酯钠、酯表面活性剂(优选spantm和/或tweentm山梨聚糖酯)以及阳离子表面活性剂(尤其是十六烷基三甲基溴化铵-ctab)和它们的混合物,b)至少一种非水性溶剂,选自二氯甲烷、氯仿、乙醇、丙酮和它们的混合物,c)超过0.1%的至少一种不溶于水的抗病毒药物,d)聚合物,选自聚乙二醇(peg)、聚乙烯醇(pva)、聚乙烯吡咯烷酮(pvp)、羟丙基纤维素(hpc)和羟丙基甲基纤维素(hpmc)、藻酸盐和它们的混合物,以及e)可选的水性介质,尤其是水。用于本发明的干燥原料是乳液或溶液,优选不含固体物质以及特别优选不含任何未溶解的抗病毒药物。特别优选在组合物中抗病毒药物的量应该使得在干燥的组合物中的负载大于或等于30%,优选大于或等于40%以及最优选大于或等于50%。这种组合物具有如上讨论的小粒径和高效用的益处。没有很好的了解在干燥步骤之后获得的材料的结构。由于不溶于水的材料的不连续的宏观体在干燥产品中不以核-壳形态存在,因此据认为所得的干燥粉末材料没有封装。干燥材料既不是‘干燥乳液’也没有易挥发的溶剂,易挥发的溶剂包括在干燥步骤之后残留的乳液的‘油相’。因为乳液可能具有‘干燥乳液’,因此添加水到干燥产品不能重组乳液。因为能够不损失益处地改变本发明存在的组分的比例,也认为组合物不是所谓的固体溶液。优选地,干燥步骤之后产生的组合物将包括抗病毒药物和载体材料,重量比从1:500到85:15(抗病毒药物:载体材料)到从1:100到85:15,并且进一步优选从1:500到1:1到从1:100到1:1。通过喷雾干燥和冷冻干燥能够获得典型量的30-50%左右的不溶于水的抗病毒药物和70-50%左右的载体材料。与上述方法相关的,优选的抗病毒药物和优选的载体材料如上所述并且在下文中将进一步详细描述。类似地,组合物的优选的物理性质如上所述。更多方面通过本发明的方法方面获得的产品适合用于制备治疗或预防病毒性疾病包括逆转录病毒性疾病的药剂,并且特别地,抗病毒药物为不溶于水的抗逆转录病毒药物时,制备的药剂用于hiv和aids的治疗或预防。本发明的第六方面提供制备用于治疗病毒性疾病包括逆转录病毒性疾病的药剂的方法,该方法包括制备根据本发明的组合物的步骤。本发明的第七方面提供一种如本文所述的组合物,该组合物用于在受试者中治疗和/或预防病毒性疾病或症状,包括逆转录病毒性疾病或症状(例如hiv和/或aids)。本发明的第八方面提供本文所述的组合物在制造药剂中的应用,该药剂用于在受试者中治疗和/或预防病毒性紊乱或症状,包括逆转录病毒性紊乱或症状,例如在受试者中用于治疗和/或预防逆转录病毒性疾病或症状(例如hiv和/或aids)。本发明的第九方面提供在需要的受试者中治疗和/或预防病毒性紊乱或症状包括逆转录病毒性紊乱或症状(例如hiv和/或aids)的方法,该方法包括向所述受试者给药治疗有效量的本文所述的组合物。本发明的第十方面提供一种用于向受试者给药不溶于水的抗病毒药物的试剂盒,该试剂盒含有本文所述的组合物。给药本发明的组合物的“受试者”为动物,尤其是温血动物,例如家畜或人,特别是人。为了更好的理解,本发明现在将仅通过不受限的实施例的方式更具体地描述。实施例下面每个实施例都是在环境温度和压力下进行的,除非另有说明。纳米分散体的制备实施例1将0.70g的hpmc和0.10g的pluronictmf127表面活性剂溶解在30ml的去离子水中;在加入30ml的乙醇后,将0.20g的甲磺酸沙奎那韦添加到水溶液中。得到清澈的乙醇/水溶液。乙醇/水溶液使用布琪微型b290(buchiminib290)喷雾干燥器在120℃以2.5ml/分钟的液体进料速率进行喷雾干燥。获得和收集含有20重量%的甲磺酸沙奎那韦的自由流动的白色粉末。将粉末分散在去离子水(5mg/ml)中用于粒径分析(malvernnanons)。甲磺酸沙奎那韦的z-均粒径在这种分散体中为430±9nm。实施例2将0.70g的hpc和0.10g的pluronictmf127表面活性剂溶解在30ml的去离子水中;在加入30ml的乙醇后,将0.20g的甲磺酸沙奎那韦添加到水溶液中。得到清澈的乙醇/水溶液。乙醇/水溶液使用布琪微型b290喷雾干燥器在120℃以2.5ml/分钟的液体进料速率进行喷雾干燥。获得和收集含有20重量%的甲磺酸沙奎那韦的自由流动的白色粉末。将粉末分散在去离子水(5mg/ml)中用于粒径分析(malvernnanons)。甲磺酸沙奎那韦的z-均粒径在这种分散体中为117±5nm。实施例3将0.70g的pvpk30和0.10g的pluronictmf127表面活性剂溶解在30ml的去离子水中;在加入30ml的乙醇后,将0.20g的甲磺酸沙奎那韦添加到水溶液中。得到清澈的乙醇/水溶液。乙醇/水溶液使用布琪微型b290喷雾干燥器在120℃以2.5ml/分钟的液体进料速率进行喷雾干燥。获得和收集含有20重量%的甲磺酸沙奎那韦的自由流动的白色粉末。将粉末分散在去离子水(5mg/ml)中用于粒径分析(malvernnanons)。甲磺酸沙奎那韦的z-均粒径在这种分散体中为74±8nm。实施例4将0.65g的hpmc和0.10g的pluronictmf127表面活性剂溶解在30ml的去离子水中。将0.05g的卵磷脂溶解在40ml的乙醇中,并在加入该40ml乙醇溶液后,将0.20g的甲磺酸沙奎那韦添加到水溶液中。得到清澈的乙醇/水溶液。乙醇/水溶液使用布琪微型b290喷雾干燥器在120℃以2.5ml/分钟的液体进料速率进行喷雾干燥。获得和收集含有20重量%的甲磺酸沙奎那韦的自由流动的白色粉末。将粉末分散在去离子水(5mg/ml)中用于粒径分析(malvernnanons)。甲磺酸沙奎那韦的z-均粒径在这种分散体中为196±2nm。实施例5将0.65g的pva和0.10g的pluronictmf127表面活性剂溶解在30ml的去离子水中,将0.05g的span80溶解在30ml的乙醇中,并在加入该30ml乙醇溶液后,将0.20g的甲磺酸沙奎那韦添加到水溶液中。得到清澈的乙醇/水溶液。乙醇/水溶液使用布琪微型b290喷雾干燥器在120℃以2.5ml/分钟的液体进料速率进行喷雾干燥。获得和收集含有20重量%的甲磺酸沙奎那韦的自由流动的白色粉末。将粉末分散在去离子水(5mg/ml)中用于粒径分析(malvernnanons)。甲磺酸沙奎那韦的z-均粒径在这种分散体中为336±7nm。上述实施例1-5制备的纳米分散体的概要示于下表1。表1实施例12345%甲磺酸沙奎那韦2020202020%hpmc7000650%pvp007000%hpc070000%pva000065%pluronictmf1271010101010%卵磷脂s7500050%span8000005z-均粒径(nm)43011774196336为了确定对细胞转移通道(transcellularpassage)的影响,评价实施例1-5的五个纳米分散体穿过caco-2细胞单层的渗透性,下文将作更详细的描述。体外肠壁渗透模型在达尔伯克改良伊格尔培养基(dulbecco'smodifiedeagle'smedium)中培养caco-2细胞并在37℃在10%的co2的孵化器中保持。融合之后,使用0.25%胰岛素-edta分离细胞,以0.5×106细胞/cm2的密度覆到聚碳酸酯膜转移小室(transwells)(0.4μm,4.7cm2生长面积,24mm插入直径)。然后每2到3天更换培养基,并且接种14-21天后细胞用于运输实验。实验之前,使用细胞电阻仪(millicell-ers)(电阻系统)测定穿过细胞单层的跨膜电阻(ter)以评价细胞单层完整性。然后在温暖的(37℃)运输培养基(hanks,含有10mmhepes的平衡的盐溶液并用氢氧化钠缓冲到ph7.4)中平衡细胞单层。去除顶端(从顶端向基底运输)或基底(从基底向顶端运输)室中的培养基并用相同体积的预暖的含有纳米颗粒或未改性的沙奎那韦的运输培养基替代。在30、60、90、120和150分钟,从接收室取得100μl样品并使用hplcms/ms分析。最后,使用放射性标记的甘露醇再评价单层的完整性,已知没有穿过完整的单层。从每时间运输的化合物的量来确定表观渗透系数(papp(cms-1))。根据下式计算papp:papp=(dq/dt)·(1/(ac0))其中:(dq/dt)为恒稳态流量(mmols-1)a为滤波器的表面积(cm2)c0为供体室的初始浓度(mm)。图1-6显示相对时间(在x轴以分钟计)转移小室中顶端侧(下面的线)相对于基底侧(上面的线)的药物浓度(在y轴以微摩尔计)。图1显示当沙奎那韦溶解在dmso中时获得的数据,作为对比例,并且图2-6显示当沙奎那韦分别根据实施例1-5以纳米分散形式制备时获得的数据。因此图1-6显示沙奎那韦以纳米微粒形式的运输与在dmso中分子状态溶解的沙奎那韦的运输一样有效。从获得的数据计算从顶端侧到基底侧的表观渗透率(pappa-b)相对于从基底侧到顶端侧的表观渗透率(pappb-a)的比率并显示在图7中。如果这种比率小于1则药物从肠到血流的运输是差的。理想地大于1的比率说明成功运输药物到血流;如果这是大量的则是好的运输。对于大多数的组合物(即,实施例1-4),基底运输似乎与正常的‘溶解的’沙奎那韦的运输(即,对比例)一样。因此,在实施例1-4中纳米分散的颗粒配方都与溶解在dmso中的非分散的沙奎那韦表现类似。对于实施例5的配方,基底运输似乎小于正常的沙奎那韦的运输(如通过更高比率显示)。然而,数量上,这种变化等于实施例5的表观渗透率增加至少6倍,并且它说明实施例5的纳米分散的沙奎那韦从顶端膜以某种方式避开流出。实施例6将1mg的依法韦仑溶解在0.1ml的氯仿中;将3mg的brij-58表面活性剂和6mg聚乙烯吡咯烷酮溶解在0.4ml的水中。结合这些溶液的每种并使用up400s超声破碎器(20%功率设定;100%负载循环7秒)乳化。所得乳液在液氮中冷冻并使用virtis台式冷冻干燥器在40微巴的恒压下冷冻干燥超过48小时。添加1ml的水到所得粉末中,并且使用dls(malvernnanons)分析所得分散体的粒径。依法韦仑的z-均粒径在这种分散体中为240nm。实施例7将1mg的洛匹那韦溶解在0.1ml的氯仿中;将3mg的多库酯钠表面活性剂和6mg羟丙纤维素溶解在0.4ml的水中。结合这些溶液的每种并使用up400s超声破碎器(20%功率设定;100%负载循环7秒)乳化。所得乳液在液氮中冷冻并使用virtis台式冷冻干燥器在40微巴的恒压下冷冻干燥超过48小时。添加1ml的水到所得粉末中,并且使用dls(malvernnanons)分析所得分散体的粒径。依法韦仑的z-均粒径在这种分散体中为423nm。实施例8将1mg的利托那韦溶解在0.1ml的氯仿中;将3mg的tween-80tm表面活性剂和6mg聚乙烯醇溶解在0.4ml的水中。结合这些溶液的每种并使用up400s超声破碎器(20%功率设定;100%负载循环7秒)乳化。所得乳液在液氮中冷冻并使用virtis台式冷冻干燥器在40微巴的恒压下冷冻干燥超过48小时。添加1ml的水到所得粉末中,并且使用dls(malvernnanons)分析所得分散体的粒径。依法韦仑的z-均粒径在这种分散体中为326nm。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1