一种镁合金增强化学转化膜的制备方法与流程
本发明涉及镁合金表面处理领域,特别是一种镁合金增强化学转化膜的制备方法。
背景技术:
化学转化膜技术是提高镁合金耐蚀性能的有效方法,但是由于在化学反应过程中化学溶液与镁合金表面原子化学反应产物的多样性产生的应力以及化学转化膜的强韧性较低,导致化学转化膜存在大量微裂纹,难以满足工程要求。
为解决化学溶液与镁合金表面原子化学反应产物的多样性产生转化膜微裂纹的问题,可在镁合金化学转化处理过程中外加磁场使溶液反应离在Lorentz力作用下作螺旋定向运动,以达获得致密均一的镁合金转化膜的目的(如国家发明专利201010217144.4“一种制备镁合金表面转化耐蚀膜层的方法”和国家发明专利201010217151.4“一种制备镁合金表面转化耐蚀膜层的装置”),也可通过在化学转化液中加入一定量的纳米颗粒促进转化膜形核,同时通过对裂纹扩展的钉扎作用抑制大量裂纹的产生(如国家发明专利201210317304.1“一种纳米氧化铝颗粒增强转化膜制备方法”和国家发明专利201210316973.7“一种给纳米颗粒加电荷的装置”)。尽管上述方法能够有效地抑制镁合金化学转化膜微裂纹的产生,但制备出的镁合金化学转化膜的耐磨性能仍然较低,不适应制造机械运动构件的要求。
技术实现要素:
为了克服现有技术的不足,本发明提供了一种镁合金增强化学转化膜的制备方法,该制备方法包括Fe3O4@NdFeB@非晶SiO2三层核壳结构磁性纳米颗粒的制备以及镁合金增强转化膜的制备,其Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜微裂纹区域生长不仅堵塞了转化膜裂纹区,而且形成转化膜的增强骨架,有效提高了镁合金转化膜的耐蚀性能和耐磨性能。
本发明解决其技术问题所采用的技术方案是:一种镁合金增强化学转化膜的制备方法,由以下制备步骤组成:
步骤一:Fe3O4@NdFeB核壳结构颗粒的制备;
步骤二:Fe3O4@NdFeB核壳结构表面非晶SiO2外壳的制备;
步骤三:镁合金的化学转化处理;
步骤四:Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜裂纹区可控生长。
优选的,所述步骤一具体为:
利用分子束外延设备(MBE)在Fe3O4纳米颗粒表面分子束外延生长NdFeB壳层:将纯度为99.99%的Nd、Fe和B作为蒸发源,其中Nd、Fe和B摩尔质量比为1-2:5-10:0.5-2,将Fe3O4纳米颗粒平铺于Si晶片上并用PTFE透气膜覆盖封闭作为外延生长衬底,对分子外延真空生长室抽真空至5×10-7-1×10-6Pa,将衬底加热至450-600℃,分别控制蒸发源Nd、Fe和B的温度为900-1200℃、700-1050℃和800-1300℃,Nd、Fe和B的蒸发束流分别为2×10-4-5×10-4Pa、6×10-4-8×10-4Pa和8×10-5-1×10-4Pa,NdFeB壳层的生长速度控制在400nm/h-800nm/h,外延生长时间为0.5-1.5小时后形成Fe3O4@NdFeB核壳结构纳米颗粒。
优选的,所述步骤二具体为:
将无水乙醇与丙酮按体积比为1:1的比例充分混合成混合溶剂,将Fe3O4@NdFeB核壳结构纳米颗粒加入上述混合溶剂,Fe3O4@NdFeB核壳结构纳米颗粒的重量百分比为12%-45%,在室温下以超声波分散20-50分钟形成稳定的Fe3O4@NdFeB核壳结构纳米颗粒分散体系,加入硅酸四乙酯至其浓度达到180-320mL/L,加入4-甲氧基苯甲醇至其浓度达到2-12g/L,加入乙酰胺吡咯烷酮至其浓度达到15-22g/L形成准备溶液。用去离子水配制体积百分比为20-75%的碳酸氢铵溶液,将配制的200-600mL碳酸氢铵溶液与1L上述配制的准备溶液充分混合,使该混合体系操持2.5-4小时;过滤得该体系固态产物,在温度为140-160℃下保温0.5-1小时以在Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳,即获得Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
优选的,还包括后处理步骤:以去离子水配制浓度为5-18mL/L的氢氟酸,将上述负载了Fe3O4@NdFeB@非晶SiO2结构的纳米颗粒浸入配制的氢氟酸溶液中,浸蚀10-25分钟以达到Fe3O4@NdFeB核壳结构表面形成的非晶SiO2外壳的局部去除,从而获得非晶SiO2层具有更高表面积的Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
优选的,所述步骤三具体为:
将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵依次加入去离子水中,形成环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵浓度分别为120-260mL/L、45-130g/L、210-340g/L、16-35g/L和5-21g/L的转化液。
将镁合金浸入转化液1-8分钟进行化学转化处理,温度为35-75℃,转化处理过程进行充分的机械搅拌,待转化处理完成后,镁合金放入40℃烘箱中持续烘干40分钟。
优选的,所述步骤四具体为:
将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒加入去离子水中,形成环己六醇磷酸酯、柠檬酸、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒浓度分别为10-35mL/L、10-75g/L和135-160g/L的电沉积液。将化学转化处理完成的镁合金作为阴极,铂为阳极,以电压幅值为5-10V,频率为50-100Hz的正弦交流电进行电沉积3-10分钟,从而在镁合金表面获得Fe3O4@NdFeB@非晶SiO2结构颗粒增强化学转化膜。
本发明的积极效果:首先本发明方法制备的Fe3O4@NdFeB@非晶SiO2结构纳米颗粒,充分发挥了NdFeB极高磁能积与Fe3O4的结合具有较高磁性的特点,且避免了NdFeB耐蚀性较差的弱点。以这种非晶SiO2外壳包覆的高磁性三层核壳结构纳米颗粒在电沉积液中对镁合金化学转化膜进行交流电沉积,镁合金转化膜微裂纹区在强烈的交流电流作用下,制备的Fe3O4@NdFeB@非晶SiO2结构磁性纳米颗粒在镁合金化学转化膜的微裂纹区域沉积生长,Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜微裂纹区域生长不仅堵塞了转化膜裂纹区,而且形成了转化膜的增强骨架,有效提高了镁合金转化膜的耐蚀性能和耐磨性能。
附图说明
图1是本发明所述Fe3O4@NdFeB@非晶SiO2结构纳米颗粒的制备流程示意图;
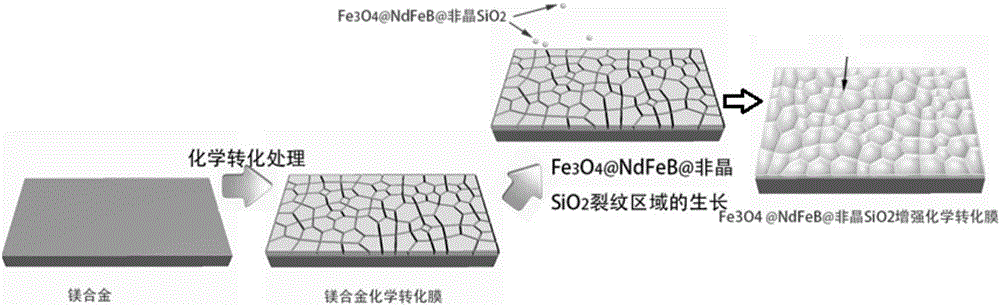
图2是本发明所述增强化学转化膜的制备流程示意图;
图3是本发明所述增强化学转化膜的结构示意图;
图4是本发明对比例、实施例1和实施例2的腐蚀电位、腐蚀电流及磨损量的实验结果图。
具体实施方式
下面结合附图对本发明的优选实施例进行详细说明。
参照图1至图3,本发明优选实施例提供一种镁合金增强化学转化膜的制备方法,按下列步骤顺序进行:
(1)Fe3O4@NdFeB核壳结构颗粒的制备:利用分子束外延设备(MBE)在Fe3O4纳米颗粒表面分子束外延长NdFeB壳层。具体为:将纯度为99.99%的Nd、Fe和B作为蒸发源,Nd、Fe和B摩尔质量比为1-2:5-10:0.5-2,将Fe3O4纳米颗粒平铺于Si晶片上并用PTFE透气膜覆盖封闭作为外延生长衬底,PTFE透气膜能够使分子束设备抽真空达到所要求的真空度,同时防止Fe3O4纳米颗粒冲出透气膜污染分子外延真空生长室,且能透过蒸发的Nd、Fe和B离子。对分子外延真空生长室抽真空至5×10-7-1×10-6Pa,将衬底加热至450-600℃,蒸发源Nd、Fe和B温度控制分别为控制在900-1200℃、700-1050℃和800-1300℃,Nd、Fe和B的蒸发束流分别为2×10-4-5×10-4Pa、6×10-4-8×10-4Pa和8×10-5-1×10-4Pa,NdFeB壳层的生长速度控制在400nm/h-800nm/h,外延生长时间为0.5-1.5小时后形成Fe3O4@NdFeB核壳结构。
(2)Fe3O4@NdFeB核壳结构表面非晶SiO2外壳的制备:无水乙醇与丙酮按体积比1:1充分混合成混合溶剂,将Fe3O4@NdFeB核壳结构纳米颗粒加入无水乙醇与丙酮的混合溶剂中,Fe3O4@NdFeB核壳结构纳米颗粒重量的百分比为12%-45%,在室温下以超声波分散20-50分钟形成稳定的Fe3O4@NdFeB核壳结构纳米颗粒分散体系,加入硅酸四乙酯使其浓度达到180-320mL/L,加入4-甲氧基苯甲醇使其浓度达到2-12g/L,加入乙酰胺吡咯烷酮使其浓度达到15-22g/L形成准备溶液。用去离子水配制体积百分比浓度为20%-75%的碳酸氢铵溶液,将配制的200-600mL碳酸氢铵溶液与1L配制的准备溶液充分混合,使该混合体系操持2.5-4小时;过滤后得该体系固态产物,在温度为140-160℃下保温0.5-1小时,以在Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳,即是Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
以去离子水配制浓度为5mL/L-18mL/L的氢氟酸,将负载了Fe3O4@NdFeB@非晶SiO2结构的纳米颗粒浸入上述配制的氢氟酸溶液,浸蚀10-25分钟以达到Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳的局部去除,从而获得非晶SiO2层具有更高表面积的Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
(3)镁合金的化学转化处理:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵依次加入去离子水中,形成环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵浓度分别为120mL/L-260mL/L、45g/L-130g/L、210g/L-340g/L、16g/L-35g/L和5g/L-21g/L的转化液。
将镁合金浸入转化液1-8分钟进行化学转化处理,温度为35-75℃,转化处理过程进行充分的机械搅拌,待转化处理完成后,镁合金放入40℃烘箱中持续烘干40分钟。
(4)Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜裂纹区可控生长:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒加入去离子水中,形成环己六醇磷酸酯、柠檬酸、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒浓度分别为10mL/L-35mL/L、10g/L-75g/L和135g/L-160g/L的电沉积液。将化学转化处理完成的镁合金作为阴极,铂为阳极,以电压幅值为5-10V,频率为50-100Hz的正弦交流电进行电沉积3-10分钟,从而在镁合金表面获得Fe3O4@NdFeB@非晶SiO2结构颗粒增强化学转化膜。
下面给出具体对比例以及实施例:
对比例:
本发明对比例提供一种镁合金转化膜方法,其制备过程为:
将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵依次加入去离子水中,形成环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵浓度分别为130mL/L、55g/L、260g/L、20g/L和7g/L的转化液。
将镁合金浸入转化液4分钟进行化学转化处理,温度为40℃,转化处理过程进行充分的机械搅拌,待转化处理完成后,镁合金放入40℃烘箱中持续烘干40分钟。
实施例1:
本发明优选实施例1提供一种镁合金增强化学转化膜的制备方法,按下列步骤顺序进行:
(1)Fe3O4@NdFeB核壳结构颗粒的制备:利用分子束外延设备(MBE)在Fe3O4纳米颗粒表面分子束外延长NdFeB壳层,具体为:将纯度为99.99%的Nd、Fe和B作为蒸发源,Nd、Fe和B摩尔质量比为1:5:1;将Fe3O4纳米颗粒平铺于Si晶片上并用PTFE透气膜覆盖封闭作为外延生长衬底,PTFE透气膜能够使分子束设备抽真空达到所要求的真空度,同时防止Fe3O4纳米颗粒冲出透气膜污染分子外延真空生长室,且能透过蒸发的Nd、Fe和B离子。对分子外延真空生长室抽真空至6×10-7,将衬底加热450℃,蒸发源Nd、Fe和B温度控制分别为控制900℃、800℃和1300℃,Nd、Fe和B的蒸发束流分别为2×10-4Pa、7×10-4Pa和8×10-5Pa,NdFeB壳层的生长速度控制在500nm/h,外延生长时间为1小时后形成Fe3O4@NdFeB核壳结构。
(2)Fe3O4@NdFeB核壳结构表面非晶SiO2外壳的制备:无水乙醇与丙酮按体积比1:1充分混合成有机混合溶剂,将Fe3O4@NdFeB核壳结构纳米颗粒加入无水乙醇与丙酮有机混合溶剂中,Fe3O4@NdFeB核壳结构纳米颗粒重量的百分比为25%,在室温下以超声波分散20-50分钟形成稳定的Fe3O4@NdFeB核壳结构纳米颗粒分散体系,加入硅酸四乙酯使其浓度达到280mL/L,加入4-甲氧基苯甲醇使其浓度达到10g/L,加入乙酰胺吡咯烷酮使其浓度达到15g/L形成准备溶液。用去离子水配制体积百分比浓度为25%的碳酸氢铵溶液,将配制的300mL碳酸氢铵溶液与1L上述配制的准备溶液充分混合,使该混合体系操持3小时;过滤该体系固态产物,在温度为150℃保温1小时在Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳,即获得Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
以去离子水配制浓度为9mL/LmL/L的氢氟酸,将负载了Fe3O4@NdFeB@非晶SiO2结构的纳米颗粒浸入上述配制的氢氟酸溶液,浸蚀15分钟以达到Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳的局部去除,从而获得非晶SiO2层具有更高表面积的Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
(3)镁合金的化学转化处理:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵依次加入去离子水中,形成环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵浓度分别为130mL/L、55g/L、260g/L、20g/L和7g/L的转化液。
将镁合金浸入转化液4分钟进行化学转化处理,温度为40℃,转化处理过程进行充分的机械搅拌,待转化处理完成后,镁合金放入40℃烘箱中持续烘干40分钟。
(4)Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜裂纹区可控生长:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒加入去离子水中,形成环己六醇磷酸酯、柠檬酸、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒浓度分别为12mL/L、15g/L和155g/L的电沉积液。将化学转化处理完成的镁合金作为阴极,铂为阳极,以电压幅值为8V,频率为50Hz的正弦交流电进行电沉积7分钟,从而在镁合金表面获得Fe3O4@NdFeB@非晶SiO2结构颗粒增强化学转化膜。
实施例2:
本发明优选实施例2提供一种镁合金增强化学转化膜的制备方法,按下列步骤顺序进行:
(1)Fe3O4@NdFeB核壳结构颗粒的制备:利用分子束外延设备(MBE)在Fe3O4纳米颗粒表面分子束外延长NdFeB壳层。具体为:将纯度为99.99%的Nd、Fe和B作为蒸发源,Nd、Fe和B摩尔质量比为1.5:7:2,将Fe3O4纳米颗粒平铺于Si晶片上并用PTFE透气膜覆盖封闭作为外延生长衬底,PTFE透气膜能够使分子束设备抽真空达到所要求的真空度,同时防止Fe3O4纳米颗粒冲出透气膜污染分子外延真空生长室,且能透过蒸发的Nd、Fe和B离子。对分子外延真空生长室抽真空至1×10-6Pa,将衬底加热至600℃,蒸发源Nd、Fe和B温度控制分别为控制在1000℃、1050℃和1200℃,Nd、Fe和B的蒸发束流分别为4×10-4Pa、8×10-4Pa和1×10-4Pa,NdFeB壳层的生长速度控制在800nm/h,外延生长时间为1.5小时后形成Fe3O4@NdFeB核壳结构纳米颗粒。
(2)Fe3O4@NdFeB核壳结构表面非晶SiO2外壳的制备:无水乙醇与丙酮按体积比1:1充分混合成有机混合溶剂,将Fe3O4@NdFeB核壳结构纳米颗粒加入无水乙醇与丙酮有机混合溶剂,Fe3O4@NdFeB核壳结构纳米颗粒重量的百分比为40%,在室温下以超声波分散20-50分钟形成稳定的Fe3O4@NdFeB核壳结构纳米颗粒分散体系,加入硅酸四乙酯使其浓度达到300mL/L,加入4-甲氧基苯甲醇使其浓度达到8g/L,加入乙酰胺吡咯烷酮使其浓度达到20g/L形成准备溶液。用去离子水配制体积百分比浓度为30%的碳酸氢铵溶液,将配制的500mL碳酸氢铵溶液与1L上述配制的准备溶液充分混合,使该混合体系操持4小时;过滤后获得该体系固态产物,在温度为160℃下保温0.5小时,以在Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳,即获得Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
以去离子水配制浓度为12mL/L的氢氟酸,将负载了Fe3O4@NdFeB@非晶SiO2结构的纳米颗粒浸入上述配制的氢氟酸溶液,浸蚀10分钟以达到Fe3O4@NdFeB核壳结构表面形成非晶SiO2外壳的局部去除,从而获得非晶SiO2层具有更高表面积的Fe3O4@NdFeB@非晶SiO2结构纳米颗粒。
(3)镁合金的化学转化处理:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵依次加入去离子水中,形成环己六醇磷酸酯、柠檬酸、氯化钙、磷酸钠和硼酸铵浓度分别为160mL/L、80g/L、220g/L、30g/L和12g/L的转化液。
将镁合金浸入转化液5分钟进行化学转化处理,温度为55℃,转化处理过程进行充分的机械搅拌,待转化处理完成后,镁合金放入40℃烘箱中持续烘干40分钟。
(4)Fe3O4@NdFeB@非晶SiO2结构纳米颗粒在转化膜裂纹区可控生长:将浓度为分析纯的环己六醇磷酸酯、柠檬酸、氯化钙、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒加入去离子水中,形成环己六醇磷酸酯、柠檬酸、Fe3O4@NdFeB@非晶SiO2结构纳米颗粒浓度分别为15mL/L、20g/L和140g/L的电沉积液。将化学转化处理完成的镁合金作为阴极,铂为阳极,以电压幅值为6V,频率为70Hz的正弦交流电进行电沉积10分钟,从而在镁合金表面获得Fe3O4@NdFeB@非晶SiO2结构颗粒增强化学转化膜。
配制5%的NaCl溶液,将对比例、实施例1和实施例2分别浸入上述配制5%NaCl溶液静置10分钟,测量对比例、实施例1和实施例2的腐蚀极化曲线;此外,将对比例、实施例1和实施例2在磨损实验机上进行磨损实验,载荷为1kg,转速为100转/分钟,磨损时间为20分钟。
根据极化曲线得到对比例、实施例1和实施例2在5%NaCl溶液中的腐蚀电位和腐蚀电流如图4所示。从图4可知,实施例1和实施例2的腐蚀电位和腐蚀电流大体相同,实施例1和实施例2的腐蚀电位明显高于对比例,而腐蚀电流明显低于对比例。从磨损实验的结果可知,实施例1和实施例2在上述磨损实验的磨损量只有对比例的1%。实验结果表明,依据本发明制备的镁合金Fe3O4@NdFeB@非晶SiO2结构颗粒增强化学转化膜有效提高了镁合金转化膜的耐蚀性能和耐磨性能。
以上所述的仅为本发明的优选实施例,所应理解的是,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想,并不用于限定本发明的保护范围,凡在本发明的思想和原则之内所做的任何修改、等同替换等等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!