具有优选形态的含杂质阴极材料及自含杂质金属碳酸盐的制备方法与流程
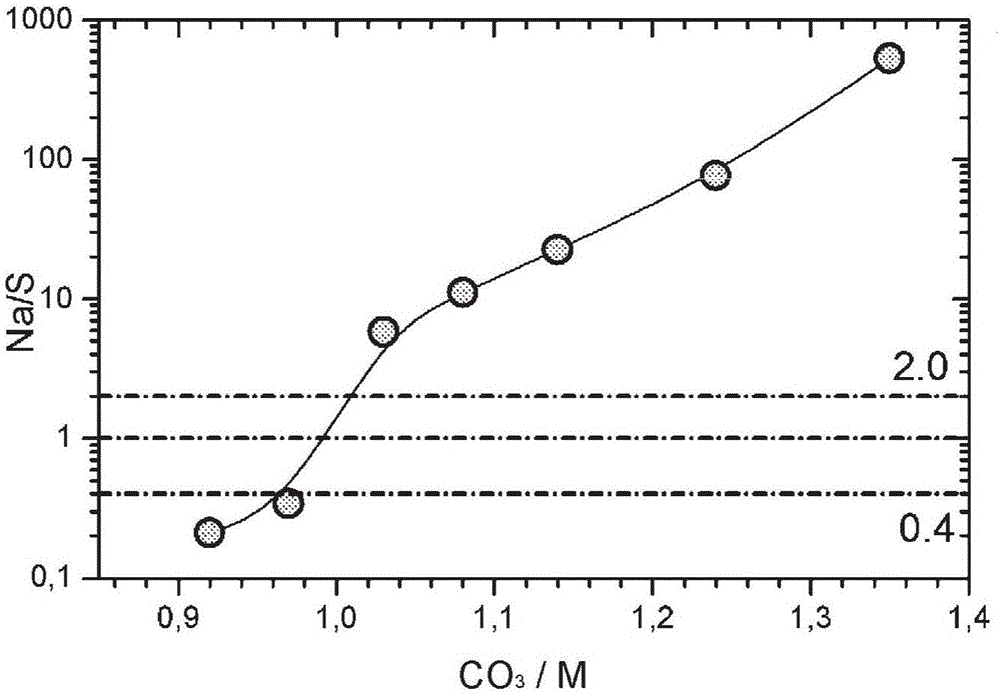
本发明涉及用于可充电锂电池的阴极材料的前驱物及生产这些前驱物的方法。此类阴极材料为所谓的NMC阴极材料,其中NMC代表镍锰钴。更特定而言,本发明着重在供应用于NMC阴极材料的前驱物,并且目标在于最终NMC阴极材料具有大表面积,因而特别适用于对电力有高度需求的应用,像是混合动力电动车的电池。NMC阴极材料一般通过固态反应制备,其中锂源(例如Li2CO3)与含Ni-Mn-Co的前驱物共混,然后将混合物在含氧氛围(例如空气)中烧制,以产生最终锂过渡金属氧化物粉末。一般而言,NMC具有的化学计量约略为LiMO2,其中M为主要由Ni、Mn及Co所组成的过渡金属。其晶体结构为有序的岩盐结构,其中阳离子会依序排成2维Li和M层。其空间群为R-3M。有许多不同的可能组成,通常依其镍、锰及钴的含量来分类及命名。典型NMC基材料为“111”(其中M=Ni1/3Mn1/3Co1/3)、“552”(其中M=Ni0.423Mn0.423Co0.167)、“532”(其中M=Ni0.5Mn0.3Co0.2)、“622”(其中M=Ni0.6Mn0.2Co0.2)、“261”(其中M=Ni0.222Mn0.667Co0.111)等。在本文件中为简洁起见,通常使用这些编号来指称金属组成,例如我们会将M=Ni0.423Mn0.423Co0.167称为M=NMC552。附加掺杂是可能的,典型元素包括Al、Mg等。一般而言,Li对M的化学计量比接近一,但通常不会刚好为一。如果Li:M增加,则Li会取代M层位点的M,其结构可以简化方式写成Li1[M1-xLix]O2或Li1+xM1-xO2,其中Li:M=(1+x)/(1-x)。“111”及“442”的典型Li:M为约1.10,而“622”的典型Li:M为1.02。提高Li:M化学计量比的一个效果是阳离子混合改变。所谓阳离子混合指的是实际晶体结构并非恰好是LiMO2或Li1[M1-xLix]O2,而是{Li1-xMx}[M1-yLiy]O2,其中“x”是指Li层位点上的M原子,其因而进行了“阳离子混合”。NMC是“混合金属”阴极材料,已知NMC无法由“非混合”前驱物制备。使用非混合前驱物(例如NiO+Mn2CO3+Co3O4)通常导致性能不佳的电极材料。为了使阴极在电池内运作良好,在Li-M-O2晶体结构中,Ni、Mn、Co阳离子需要在原子尺度上充分混合。在标准制程中,这是通过使用混合过渡金属前驱物来达成,其中过渡金属原子在纳米尺度上充分混合。针对NMC阴极的制备,通常使用混合金属氢氧化物M(OH)2、或其氧化形式的MOOH作为前驱物。混合氢氧化物前驱物通常通过沉淀过程制备。工业上广泛使用的制程包括以下步骤:将(a)金属硫酸盐溶液、(b)NaOH溶液及(c)NH4OH溶液的流馈入反应器中。所得氢氧化物含有硫,但实际上不含钠。大部分的硫在前驱物烧制期间仍残留,因此最终市售NMC阴极含有硫。制备混合氢氧化物前驱物的标准沉淀过程涉及氨的使用。氨为所谓的螯合剂。Ni-氨络合物会提高金属溶解度,因而降低沉淀期间的成核率。例如,如果不使用氨,则难以制备出足够致密的氢氧化物,尤其是如果粒径>10μm的大粒子为所期望的时。如果不使用氨,则实际上不可能稳定过渡金属氢氧化物的沉淀条件,即无法产生具有优选球形形态的大粒子。存在于沉淀过程的氨总是产生某些安全风险。如果发生意外,有害烟雾会逐渐形成,所以就安全性的观点来看,开发无氨沉淀过程是高度理想的。在沉淀之后,氨残留于过滤溶液。由于氨不得被排放至环境中,因此废水要经过处理以去除(优选地回收)氨。这些除氨装置相当昂贵,显著增加资本投资,同时特别是因为对能源的较高需求而增加废料处理的操作成本。因此期望开发一种无氨沉淀方法,其因下述理由而供应具有足够密度及球形形态的混合前驱物。还有使用混合金属碳酸盐作为NMC前驱物的报告,但就我们所知尚未在工业上使用。用于锂过渡金属氧化物阴极材料的混合金属碳酸盐前驱物的制备长久以来为熟知的。例如,US7879266公开粒径介于20和40μm之间且布厄特(Brunauer-Emmett-Teller,BET)表面积介于50和130m2/g之间的混合金属碳酸盐前驱物。敲紧密度超过1.7g/cm3。制备使溶解的过渡金属盐与碳酸盐或碳酸氢盐溶液共沉淀。此沉淀在CO3/M比为2至10、优选3至8下发生。US7897069公开用于制备NMC的混合金属碳酸盐前驱物。其粒径为5至20μm并且BET(Brunauer-Emmett-Teller)表面积为40至80m2/g。敲紧密度超过1.7g/cm3。该制备是使溶解的过渡金属盐与碳酸盐或碳酸氢盐溶液共沉淀。此沉淀在CO3/M比为2至7、优选3至6下发生。该专利的方法在反应溶液中使用过量的碳酸盐(CO3),使其达成具有高产率的复合碳酸盐。然而,使用过量Na2CO3会使所得的碳酸盐具有大量Na杂质,并且由CO3过量前驱物制备的LiMO2阴极显示性能不佳。其他碳酸盐制程专利为CN101229928B,其描述一种包括氨的碳酸盐沉淀过程,而US8066915描述该对应制程。US7767189描述一种制备NMC的过程,其大致上包括混合金属碳酸盐的沉淀。在该碳酸盐沉淀反应中,所使用的是腐蚀性比NaOH小的Na2CO3,并且碳酸盐沉淀期间的pH较低,这表示该反应的腐蚀性比氢氧化物沉淀低。因此,碳酸盐过程较易于在量产规模上实施。其他替代沉淀方法包括碳酸氢盐沉淀。通过下列碳酸氢盐过程反应,可相对容易地达成具有所需特性(像是球形形态、良好密度等)的混合碳酸盐前驱物:2NaHCO3+MSO4→Na2SO4+MCO3+H2CO3。(1)但该过程的缺点在于效率低。要沉淀1kg的MCO3通常需要约1.5kg的碳酸氢钠,而碳酸盐过程Na2CO3+MSO4→Na2SO4+MCO3(2)所需的碳酸盐远为较少,仅约900g。此外,碳酸氢盐的溶解度(在90℃下约200g/L)远低于碳酸钠(约400g/L)。这表示相较于碳酸盐沉淀,碳酸氢盐过程每升溶液的最大产率低了3倍,这显著提高了过滤及废水处理成本,使得碳酸氢盐过程不完全具有竞争力。相较于碳酸盐沉淀,碳酸氢盐沉淀于高浓度碳酸盐存在下会在较低pH下发生。较低pH倾向于抑制Na的插入,而过量CO3倾向于抑制硫插入混合过渡金属碳酸盐晶体结构中。因此,碳酸氢盐可能使相对不佳的过渡金属碳酸盐沉淀。针对LiMO2阴极的制备,一般期望获得纯MCO3前驱物。高杂质含量倾向于降低LiMO2阴极的可逆容量,因为有电化学“惰性”第二相存在。因此趋于认为硫并不是所期望的,并且钠杂质尤其有害。但本专利申请的作者仔细研究杂质是否可被忍受或甚至为所期望的,而如果为期望的,杂质(尤其是硫及钠)存在的浓度及mol比应为多少。对于汽车应用像是混合动力电动车(HEV,HybridElectricVehicle)而言,需要较高功率的电池。阴极材料需要能够支持这些高功率倍率。倍率性能的一个主要限制是单一粒子内的锂的固态扩散率。一般而言,如果固态扩散长度缩短,则可减少典型扩散所需时间,因此可达成较高功率。扩散长度可通过缩小粒径来缩短,但由于小粒子具有低密度而仍有限制。此低密度并不是所期望的,因为这会在电极涂布期间造成问题,且最终电池的体积能量密度为低。远为优选的是相对较大、球形且相对致密的粒子,其具有开放、互连孔隙。在本文件中,我们将具有大球形、相对致密粒子,但同时具有互连中孔或纳米孔隙的NMC阴极粉末称为“具有优选形态的NMC”。开放、互连孔隙会增加表面,因此“具有优选形态的NMC”的BET表面积远比预期来自具有相同形状的致密粒子要来得高。所以由致密粒子所组成的市售NMC的BET表面积通常在0.2至0.4m2/g的级数。具有优选形态的NMC通常具有的BET表面积在1m2/g或更高的范围。在电池中,具有优选形态的NMC的孔隙将会填满电解质,而电解质会作为进入粒子的扩散快捷方式,因为液体扩散会比固体粒子中的扩散要快得多。然而,要获得具有此优选形态的粒子的粉末仍是挑战。本发明的目的在于提供NMC阴极材料及用于这些NMC阴极材料的碳酸盐基前驱物,该NMC阴极材料尤其适用于汽车应用中。技术实现要素:就第一方面观看,本发明可提供一种用于制造可在锂离子电池中用作活性正极材料的锂金属(M)氧化物粉末的碳酸盐前驱物化合物,M包括20至90mol%Ni、10至70mol%Mn及10至40mol%Co,该前驱物还包括钠及硫杂质,其中该钠对硫的摩尔比(Na/S)为0.4<Na/S<2。在一个实施方案中,该碳酸盐前驱物可具有通式MCO3,其中M=NixMnyCozAv,A为掺杂物,其中0.20≤x≤0.90,0.10≤y≤0.67,且0.10≤z≤0.40,v≤0.05,并且x+y+z+v=1。在另一实施例中,该碳酸盐前驱物化合物可具有通式MCO3,其中M=NixMnyCozAv,A为掺杂物,其中0.30≤x≤0.60,0.20≤y≤0.50,且0.10≤z≤0.40,v≤0.05,并且x+y+z+v=1。在另一个实施方案中,v=0。在又一个实施方案中,该碳酸盐前驱物化合物可具有通式MCO3,其中M=NixMnyCozAv,A为掺杂物,其中0.10≤x<0.30,0.55≤y≤0.80,而0<z≤0.30,v≤0.05,且x+y+z+v=1。在另一实施例中,v=0。在多个实施方案中,该掺杂物A可为Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn及B中的任一者或多者。针对本发明的碳酸盐前驱物化合物,以重量%表示的该钠(Na重量)及硫(S重量)含量的总和(2*Na重量)+S重量可超过0.4重量%且小于1.6重量%。在该实施方案中,钠含量可介于0.1和0.7重量%之间,并且硫含量介于0.2和0.9重量%之间。同样在该实施方案中,钠含量可介于0.15和0.30重量%之间,并且硫含量介于0.20和0.45重量%之间。该碳酸盐前驱物化合物可具有10μm≤D50≤20μm的粒径分布。本发明的第一方面为提供一种用于NMC阴极材料的混合金属碳酸盐前驱物。在烧制之后,此类NMC阴极材料具有优选的形态。所得的阴极具有开放孔隙,且BET表面积显著高于非根据本发明所制备的相同尺寸的致密粒子所预期的。该优选形态支持较高功率,使该NMC阴极适用于汽车应用。该前驱物含有钠及硫杂质,这些杂质的浓度及比例经适当设计以达到优异性能。可清楚得知,根据本发明的其它前驱物实施方案可通过结合各个前述不同前驱物实施方案中所涵盖的特征而提供。就第二方面观看,本发明可提供一种用于可充电电池中的正极材料的锂金属氧化物粉末,其具有通式Li1+aM1-aO2,其中M=NixMnyCozAv,A为掺杂物,其中-0.05≤a≤0.25,0.20≤x≤0.90,0.10≤y≤0.67,且0.10≤z≤0.40,v≤0.05,并且x+y+z+v=1,该粉末具有10μm≤D50≤20μm的粒径分布、0.9≤BET≤5的比表面,该BET以m2/g表示,该粉末还包括钠及硫杂质,其中以重量%表示的该钠(Na重量)及硫(S重量)含量的总和(2*Na重量)+S重量超过0.4重量%且小于1.6重量%,并且其中该钠对硫摩尔比(Na/S)为0.4<Na/S<2。在一个实施方案中,该锂金属氧化物粉末包括次要LiNaSO4相。在另一个实施方案中,该次要LiNaSO4相的相对重量为至少0.5重量%,如由该粉末的XRD图谱的里特韦尔(Rietveld)分析所测定的。优选的是,该相对重量为至少1.5重量%,或甚至至少3.5重量%。该掺杂物A可为Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn及B中的任一者或多者。在多个实施方案中:-0.05≤a<0.10,0.30≤x≤0.60,0.20≤y≤0.50,且0.10≤z≤0.40,或0.10≤a≤0.25,0.10≤x<0.30,0.55≤y≤0.80,且0<z≤0.30。针对本发明的锂金属氧化物粉末,其可为下列任一者:0.4<Na/S<1,且该粉末还包括Na2SO4;或1<Na/S<2,且该粉末还包括Li2SO4。本发明的第二方面为一种由该混合金属碳酸盐制备的阴极材料。该阴极材料具有优选形态。粒子一般为球形、具有开放孔隙且表面显著大于由类似形状的致密粒子所组成的阴极材料的表面。可清楚得知,根据本发明的其它锂金属氧化物粉末实施方案可通过结合各个前述不同粉末实施方案中所涵盖的特征而提供。就第三方面观看,本发明可提供一种用于制备碳酸盐前驱物化合物的方法,其包括下列步骤:-提供包含Ni离子、Mn离子、Co离子及A源的进料溶液,-提供包含碳酸盐溶液及Na离子的离子溶液,-提供包含含有M’离子的晶种的浆料,其中M’=Nix’Mny’Coz’A’n’,A’为掺杂物,并且0≤x’≤1,0≤y’≤1,0≤z’≤1,0≤n’≤1且x’+y’+z’+n’=1,-在该反应器中混合该进料溶液、该离子溶液与该浆料,从而获得反应性液体混合物,-使碳酸盐沉淀在该反应液体混合物中的此类晶种上,从而获得经反应的液体混合物与该碳酸盐前驱物,以及-从该经反应的液体混合物分离该碳酸盐前驱物。在一个实施方案中,此类晶种具有介于0.1和3μm之间的中值粒径D50。在另一个实施方案中,此类M’离子存在于水不溶性化合物中,该水不溶性化合物为M’CO3、M’(OH)2、M’氧化物及M’OOH中的任一者。此类Ni离子、Mn离子、Co离子及A离子可存在于水溶性硫酸盐化合物中。在另一个实施方案中,该晶种浆料中的金属含量对该进料溶液中的金属含量的摩尔比(M’晶种/M进料)介于0.001和0.1之间,并且该碳酸盐前驱物的中值粒径由M’晶种/M进料来确定。在特定实施方案中,M=M’。在不同方法实施方案中,A及A’为Mg、Al、Ti、Zr、Ca、Ce、Cr、Nb、Sn、Zn及B中的任一者或多者。在本发明的方法中,该反应器中的NH3浓度可低于5.0g/L,或该反应液体混合物可基本上无NH3。该反应器可为连续搅拌槽反应器(CSTR)。在另一个实施方案中,该离子溶液还包括氢氧化物溶液及碳酸氢盐溶液中的任一者或两者,并且OH/CO3比、或OH/HCO3比、或此二比均小于1/10。本发明的第三方面为一种通过连续沉淀过程制备这些混合碳酸盐前驱物的廉价制程。将经溶解的Na2CO3及金属硫酸盐的进料馈入反应器。碱:酸流速比(Na2CO3:MSO4)经控制以获得具有所期望Na及S杂质浓度比的混合金属碳酸盐。粒子的成长并非通过调控流速比来控制,而是通过将晶种加至反应器中来控制。就第四方面观看,本发明可提供一种用于制备根据本发明第二方面的锂金属氧化物粉末的方法,其包括下列步骤:-根据本发明的第一方面提供碳酸盐前驱物,-提供Li前驱物化合物,-混合该M碳酸盐及该Li前驱物,-将该混合物加热至500℃,其中在300和500℃之间的温度提高在至少1小时内进行,以及-在600和1100℃之间的温度下烧制该混合物至少1小时。可清楚得知,根据本发明的其它方法实施方案可通过结合各个前述不同方法实施方案中所涵盖的特征而提供。附图说明图1:碳酸盐沉淀的杂质分布图,其中CO3/M流比从0.92变化至1.35图2:碳酸盐沉淀的Na/S比随酸碱比的变化图,其中CO3/M流比从0.92变化至1.35图3:碳酸盐沉淀的杂质分布图,其中CO3/M流比从0.97变化至1.12图4:具有不同金属组成的含Na及硫碳酸盐沉淀的杂质分布图图5:在沉淀条件广泛变化后所获得的样本的杂质分布图图6:典型10LCSTR反应器的设计图7:溢流中粒子的粒径随沉淀时间的变化图图8:碳酸盐沉淀中D50随碱/酸流比的变化图图9:来自相同晶种量及不同CO3/M比的MCO3样本的PSD(D50,跨距)结果图10:来自相同晶种量及不同CO3/M比的MCO3样本的ICP(Na,S)结果图11:来自相同晶种量及不同CO3/M比的MCO3样本的敲紧密度(TD)结果图12:根据本发明的MCO3沉淀物的SEM剖面图13:用MCO3沉淀物制备的552NMC阴极的SEM显微图图14:552NMC阴极LX0142的剖面图15:全电池测试的倍率能力结果图16:全电池测试在室温下的循环稳定性图17:全电池测试在45℃下的循环稳定性图18:LiNaSO4的XRD粉末绕射图谱及计算图谱图19:NMC=261含钠及硫前驱物的SEM显微图图20:NMC=261含钠及硫最终产物(HLM330)的SEM显微图图21:样本HLM330的XRD粉末绕射图谱,其中y轴为对数尺度图22:样本HLM330的XRD粉末绕射图谱,其中y轴为线性尺度图23:经沉淀的含钠及硫NMC=532金属碳酸盐的SEM显微图图24:NMCEX1534的SEM显微图图25:样本EX1534的XRD粉末绕射图谱图26:样本EX1577的FESEM显微图图27:样本MX0809的FESEM显微图图28:关于干粉进料的示意模型具体实施方式混合金属碳酸盐原则上可通过将MSO4流及Na2SO4流连续注入搅拌反应器中来沉淀。作者观察到无论选择什么条件,根据上式(2)的纯混合碳酸盐不可能沉淀。钠和/或硫总是被包括在沉淀粒子中,且可能存在于混合碳酸盐的晶体结构内。可能的反应式为ANa2CO3+BMSO4→Na2SO4+{M1-2xNa2x}{(CO3)1-y(SO4)y}(3)在此式中,A及B接近1,x及y为小数字,通常小于0.05。在此式中,2x/(1-2x)=Na/M,其为沉淀物中的钠(杂质)含量,而y/(1-2x)=S/M,其为硫(杂质)含量。最终2x/y=Na/S,其为沉淀物中的钠对硫杂质比(mol/mol)。杂质总是存在的事实可能是碳酸盐沉淀一般被视为无法获得良好NMC阴极材料的制程的一个原因。作者认识到移除这些杂质并非优选选项,反之如果这些杂质的量在特定范围内,可达成优异的阴极特性。在烧制期间不仅形成锂过渡金属阴极,钠及硫还会反应。如果碳酸盐前驱物中的钠硫比大于2,则钠会结合至锂过渡金属阴极的晶体结构中,或者形成非期望的钠过渡金属氧化物。在这两种情况中,阴极材料所达成的性能不佳。然而,如果1<Na/S<2,则钠不会结合至晶体结构中,反而形成Na2SO4及LiNaSO4盐。这些可通过洗涤过程移除,然而作者意外观察到,这种盐的存在改善阴极在最终电池中的电化学性能。如果0.4<Na/S<1,则钠不会结合至晶体结构中,但形成Li2SO4及LiNaSO4。这些盐可通过洗涤过程移除,然而作者意外观察到,这种盐的存在改善阴极在最终电池中的电化学性能。如果钠硫比小于0.4,则硫杂质通常过高。这会过度减少理论可用容量,因为最终Li2SO4杂质在电化学上为“惰性”,即其不会增加电池的可逆容量。该Li2SO4可被洗掉,然而在此情况中,显著量的锂会损失在废水中,这提高了阴极的成本。一般而言,金属碳酸盐前驱物中的期望的Na/S比介于0.4和2之间,或者0.4<Na/S<2。最终阴极含有LiNaSO4次要相。如果钠硫比接近一,则LiNaSO4的含量最大化。在此情况中,LiNaSO4可通过XRD绕射检测,尤其可采用低速扫描以达到高计数。但如果钠硫比增加或减少而偏离越多时,越少的LiNaSO4存在,且通过XRD检测则变得困难;虽然LiNaSO4仍然存在。不只钠硫杂质比重要,总杂质含量还有关系。如果总杂质含量过少,则无法利用LiNaSO4及其他硫酸盐的效益,NMC阴极将有电化学性能不佳的问题,尤其是不可逆容量会增加。如果杂质含量过高,则存在过多电化学惰性钠盐,可逆容量单纯因为活性材料较少而减少。优选杂质含量范围如下:0.4重量%<2xNa(重量%)+S(重量%)<1.6重量%。本专利申请的目的在于供应同时含有钠及硫的前驱物,其中钠及硫的含量经优化以使最终NMC阴极可含有结晶LiNaSO4,这表示钠杂质以可溶性盐存在,因此具有优异的电化学性能。LiNaSO4通常源自碳酸盐前驱物中的杂质。有可能无法获得具有正确钠硫比的混合金属碳酸盐前驱物,反而获得具有钠硫比过高的碳酸盐前驱物。在此特殊情形中,最终阴极中的期望的钠硫比仍可通过添加硫源来达成。硫在烧制之前加入,例如以Li2SO4的形式。典型反应为Li2SO4+Na→LiNaSO4+Li。Na萃取自该锂过渡氧化物,且锂被插入该锂过渡氧化物晶体结构。典型金属氢氧化物沉淀为达到稳定态的连续制程。所使用的是连续流式反应器,其中溶解碱(例如NaOH)流及溶解酸(例如MSO4)流连续馈入该搅拌反应器中。在稳定态中,粒径控制典型通常通过变化流比来达成。在狭窄范围内,NaOH(碱)及MSO4(酸)之间的流比的微小变化允许达成不同的粒径。混合金属碳酸盐通过共沉淀反应来制备。将溶解Na2CO3流及至少溶解金属硫酸盐流馈入剧烈搅拌下的反应器中。金属流通常为不同过渡金属硫酸盐的混合物。另选地,金属硫酸盐可通过多个分开的进料馈入。通常,搅拌通过旋转叶轮来达成,但其他解决方案像是循环流也是可能的。沉淀反应优选为连续沉淀,其中进料注入具有溢流的反应器中并且产物从反应器连续排出。另选地,沉淀反应也可以批次过程执行。除了碳酸钠及金属硫酸盐的基础流外,可加入更多流像是金属氯化物碳酸氢钠、碳酸氢铵等。作者观察到在金属碳酸盐沉淀期间,保持粒径稳定比起氢氧化物沉淀要来得困难许多,这表示通过控制Na2CO3/MSO4碱:酸流速来执行稳定态沉淀更加困难。作者观察到在欲将要达到稳定态时导致MCO3沉淀物中有所期望的Na/S杂质比及含量的流速比,会造成非常大的粒子,通常超过30μm。对于电池应用而言这些粒子过大。本发明对此问题提供解决方案,并且将Na2CO3/MSO4比例保持在导致优选Na/S比的比例下。为了控制粒径,应用外部种晶方法。在沉淀期间加入作为晶种的适当小粒子,并且通过控制晶种添加率,可加以控制沉淀物MCO3的粒径,如共同提出的专利申请EP14188028.6中所述。该专利还提供一种用于本发明的碳酸盐前驱物的无氨沉淀过程,该过程适用于量产。在沉淀之后,沉淀物通过合适分离技术如过滤从液体分离出来。一般而言,不需要离子交换操作(例如用苛性NaOH溶液洗涤)。用苛性溶液洗涤可调整钠及硫含量。例如,苛性洗涤(通过使含杂质碳酸盐暴露于稀释碱像是NaOH中)适合用来减少硫杂质含量。在过滤或苛性洗涤之后,通常进行使用水的洗涤过程。仔细洗涤可移除存在杂质的一部分。接着在低于400℃下的典型干燥温度下干燥该碳酸盐前驱物。另选地,可在较高温度下烤干该碳酸盐。接着将该混合金属碳酸盐与Li源例如Li2CO3混合,然后在含氧氛围中烧制。因为Li、Na及硫同时存在,因此在烧结之后,LiNaSO4会存在于该锂过渡金属中。所生成产物为含钠及硫的过渡金属碳酸盐,其尤其适合作为锂过渡金属阴极材料的前驱物。此过程可能有多种变化。例如,也可将Li2CO3加至经洗涤(潮湿)滤饼,接着进行干燥或预烧制。最终阴极材料具有优选形态。粒子具有球形形状并展现开放孔隙。BET表面积大于类似形状但致密的粒子。开放孔隙与混合金属碳酸盐前驱物的使用密切相关。作者相信在烧制过程中,一个重要步骤是来自MCO3的CO2初始释放,而锂化反应会在稍微较高的温度下开始。CO2释放的反应方程式可写成MCO3→MO1+x+CO2。在x=0的情况中,5个原子(M+C+3O)里只有2个留在固体中。作者假设从粒子中心释出CO2产生了“烟囱”,最后造成最终阴极材料的开放孔隙。通过应用适合的烧制设定,最终阴极产物保留开放孔隙。如果以小规模制备阴极材料,则达成开放孔隙会相对容易。然而如果该阴极材料以工业方式烧制,例如使用托盘并且数公斤的前驱物共混物在一个托盘里烧制,则开放孔隙难以达成。作者观察到缓慢加热该共混物非常重要。尤其重要的是,托盘不得在介于300和500℃之间的温度范围内快速加热。适当的温度曲线需要至少2小时来将温度从300℃提高到500℃。如果在此温度范围内的加热速率太快,例如不到1小时就从300℃加热到500℃,则所获得的阴极材料会具有不佳性能。在本专利的实施方案中,所提供者为一种碳酸盐前驱物,其能够制备具有较高表面积及开放孔隙的NMC阴极粉末,从而使所获得的NMC阴极尤其适用于高功率应用。BET表面积是评估开放孔隙的重要工具。如果粒子致密,则表面积低,因此如果表面积显著大于给定尺寸粒子的预期表面积,则开放孔隙可能存在。许多提出制程的变化型均为可能的。本发明在下列实施例中进一步说明:实施例1:含Na及硫的碳酸盐的沉淀不同酸对碱流速比的第一系列沉淀(7个样本)执行如下:1)制备M=Ni0.42Mn0.42Co0.16(552)的混合金属硫酸盐溶液。金属浓度为2mol/升2)制备浓度为每升2molCO3的Na2CO3溶液3)将该金属硫酸盐及该碳酸盐溶液的连续流馈入剧烈搅拌(1000rpm)下的含水反应器中。将反应器保持在90℃下。总流速经选择以在2.8小时内将反应器的容积置换。碱对酸的摩尔流速比(CO3/SO4)固定在介于0.92至1.35之间的值。流速以重力方式来控制及固定。沉淀进行6小时。4)在每小时操作之后收集少测量试样本(得到样本1、2、…6)。浆料中的碳酸盐沉淀的粒径由雷射绕射检验。5)最终样本从第4小时收集到第6小时。最终样本在水中重复洗涤以去除任何残余盐;过滤并在120℃下风干6)收集滤液并进行检查,以测定任何未沉淀金属含量。并且还通过pH滴定来检验碱(Na2CO3)的过量最终样本通过XRD、BET表面积、敲紧密度测量、FESEM、ICP(测定元素Ni、Mn、Co、Na、S)来分析。表1显示所获得的最终样本结果。准确达到金属组成“552”的0.5%内(Ni=0.418,Mn=0.414,Co=0.166)。表1以及图1显示钠及硫的ICP分析结果。纯金属碳酸盐显然不会沉淀,并且在所有情况中有相对大量的硫酸盐和/或钠杂质存在。洗涤并未去除钠及硫。进行多次离子交换尝试,但钠杂质的去除尤其困难。图1显示1<Na/S<2的范围。如将在稍后所显示的,这些含钠及硫的碳酸盐为NMC型阴极材料的优异前驱物。然而,如果Na/S>2则性能极差。该图还显示0.4<Na/S<1范围,其还产生优异的最终NMC阴极材料。但如果Na/S<0.4,则硫含量过高,并且该NMC阴极的可逆容量不足。虚线显示优选范围。优选前驱物位于Na/S=2:1虚线及Na/S=0.4:1虚线之间。图2显示Na/S比随碱对酸(CO3/M)比的变化。随着流速比变化,Na/S比有巨幅改变。优选前驱物在0.4至2.0的狭窄范围内。表1:具有金属组成M=Ni0.42Mn0.42Co0.16的沉淀碳酸盐的流速比系列最终样本的化学分析结果使用M=Ni0.60Mn0.20Co0.20(622)的混合金属硫酸盐溶液来执行具有相对接近1/1比的不同碱对酸流速比的第二系列沉淀。如同第一系列的实验,最终样本通过XRD、BET表面积、敲紧密度测量、FESEM、ICP(测定元素Ni、Mn、Co、Na、S)来分析。表1.2显示所获得的最终样本结果。准确重现金属组成的0.5%内(Ni=0.6,Mn=0.2,Co=0.2)。表2以及图3显示钠及硫的ICP分析结果。在所有情况中,有相对大量的硫酸盐和/或钠杂质存在,无法沉淀出无杂质的金属碳酸盐。表2:具有金属组成M=Ni0.60Mn0.60Co0.20的沉淀碳酸盐的流速比系列最终样本的化学分析结果实施例2:具有不同金属组成的含Na及硫碳酸盐的沉淀重复实施例1的金属碳酸盐的制备,差别在于改变混合硫酸盐溶液的金属组成。针对一些组成使用数种流速比,另一些情况仅测试两种流速比。一般而言,沉淀条件经选择以接近或落入Na/S比例介于0.4至2之间的所期望范围。表3汇总了沉淀条件以及所获得的杂质。图4显示针对杂质的ICP分析结果(o=实施例1,Δ=M系列;■=例外)。显然无法沉淀不含杂质的金属碳酸盐。在所有情况中,钠和/或硫杂质均存在。图4显示1<Na/S<2的优选范围。一般而言,具有不同金属组成的碳酸盐会遵循如实施例1中552所示的趋势(还显示在图2上)。仅有少数例外(在低碱/酸(CO3/M)流速比下沉淀的无Ni化合物)具有较少杂质。这些无Ni化合物并不适合用作NMC前驱物。虚线是用来引导目光找到典型杂质。通过适当变化流速比,这些杂质可经调整以使得金属碳酸盐具有在优选范围内的组成。1<Na/S<2,分别为0.4<Na/S<1。表3:不同金属比及所选CO3/M(碱/酸)比的沉淀样本ID组成碱/酸ICPNaICPSNa/SNiMnCoMol比mol%mol%Mol比MCO-00891001.080.900.571.58MCO-00900101.082.420.2012.21MCO-00910011.080.180.141.29MCO-00921101.081.230.139.34MCO-00930111.082.540.0735.1MCO-00941011.080.330.321.02MCO-00956221.080.940.243.88MCO-00965321.081.050.176.3MCO-00972611.082.480.289.01MCO-0098227801.082.570.328.02MCO-01046221.123.630.0658.47MCO-01051001.000.651.420.46MCO-01060101.001.150.601.9MCO-01070011.003.761.532.46MCO-01081101.000.680.710.96MCO-01090111.001.300.284.63MCO-01101011.000.151.250.12MCO-01116221.000.481.030.47MCO-01125321.000.560.750.74MCO-01132611.001.610.513.13MCO-01141111.000.720.661.1MCO-01192611.031.670.473.57实施例3:沉淀条件的变化在此实施例中,沉淀条件经变化以研究Na及S杂质随CO3/M流比改变而偏离一般趋势的可能性。在一些情况中,10%的Na2CO3以2NaHCO3取代(在此情况中Na浓度固定在4mol/L;流速比限定为0.5*Na/SO4)。在一些情况中,10%的Na2CO3以NaOH替代(每1molNa2CO3以2molNaOH替代)。在一些情况中改变沉淀温度(至25℃),在一些情况中改变反应物的浓度,在一些情况中应用种晶技术,在一些情况中改变反应器的几何,在一些情况中改变滞留时间。大多数实验均使用金属组成NMC=552。结论是一般而言MCO3均含有杂质,在所有情况中都无法获得无杂质MCO3。图5汇总了结果。实施例4:对于含钠及硫过渡金属碳酸盐无法进行传统PSD控制本实施例显示难以控制金属碳酸盐沉淀物的PSD。优选沉淀过程为连续过程(还已知为连续流式反应器)。图6显示连续搅拌槽反应器(CSTR)的设计,其标号说明如下:1水夹套2溢流3给料管4马达5叶轮6pH传感器7檔板8温度传感器9出口阀替代性批式制程在量产层级上对物流管理(logistically)要求更高。图7左半部显示延长沉淀12小时、滞留时间2.7小时的结果(粒径(D50)对沉淀时间(h))。金属组成为552,碱对酸流摩尔比(CO3/M)为1.24。显然,在沉淀开始时有相对小粒子沉淀,其具有约12μm的D50。将Na2CO3及MSO4的连续流加入并使沉淀产物溢流。在沉淀期间,检测溢流的PSD。显然,D50持续成长。在12小时后沉淀可能缓慢达到稳定态,并且在此状态中D50超过30μm,这对于许多应用而言过大。作者相信对于成长过程有充分了解(成核率、通过溢流稀释晶核、成长率…);然而详细讨论成长模型超出本专利申请的范围。在将会产生具有所期望Na/S范围内的杂质前驱物的流比下重复实验,并且选用1.02的流比。图7右半部显示PSD参数随时间的变化。在1.02条件下,D50值显然还持续成长,显示难以控制PSD。如果希望获得例如10微米的D50,则无法通过“未经控制的”碳酸盐沉淀过程来达成。实施例5:碳酸盐沉淀过程中的粒径控制用于混合氢氧化物的典型沉淀过程为连续沉淀,其中粒径通过小心控制流速(酸对碱)比来调整。此方法是基于特定流速比获得特定稳定态粒径的事实。因此,如果碱/酸比增加,稳定态期间沉淀物的PSD通常减小,所以利用小幅度变化酸对碱流速比来将粒径控制在狭窄的所期望的范围。背后的科学原因是成核率对于pH的依赖性。当pH增加,成核率提高且粒径减小。此实施例将会显示对于含钠及硫混合碳酸盐而言,这一过程实际上不可行。如在本发明中,流速比由达到所期望的钠硫比的需求来确定,其根据该条件加以调整。因此无法与杂质控制无关地控制PSD。如实施例4中所示,这是因为当Na/S比是沉淀期间的决定因子时,连续沉淀期间所获得的粒径非常大。表4显示在6小时沉淀后的最终PSD参数,M为实施例1的552组成。大多数情况甚至未达到稳定态,因此如果沉淀继续,D50会进一步成长。如果所期望的PSD是10μm,则此仅可通过选择低于0.97的CO3/M来达成。然而,在这些条件下所沉淀氢氧化物的硫杂质非常高并且Na对S值小于所期望的0.4比值。图8显示D50值随流速比CO3/M的变化而增加。显然,Na/S杂质范围(参见实施例1、2)以及PSD范围(参见实施例4、5)均高度取决于相同的流速比。因此不可能沉淀具有所期望杂质含量的MCO3前驱物同时又达到所期望的粒径。表4:在沉淀6小时之后来自CO3/M系列的MCO3样本的分析结果SPAN=(D90-D10)/D50实施例6:应用种晶技术进行PSD控制如先前实施例中所示,金属碳酸盐沉淀过程(Na2CO3+MSO4→Na2SO4+MCO3)的其中一个问题是PSD控制。相对于氢氧化物沉淀的情况(其中粒径通过流速比((OH)2/M)控制),在碳酸盐沉淀的情况中,我们无法轻易生产不同粒径的金属碳酸盐前驱物,因为此沉淀过程对于流速控制远比金属氢氧化物沉淀过程更加敏感。已发现在金属碳酸盐沉淀期间应用种晶技术能够精确控制粒径,并且轻易达成稳定态过程,如在共同提出的专利申请EP14188028.6中所公开的。在一个实施方案中,该过程进行如下:晶种制备过程:步骤(1)及(2)步骤(1):金属碳酸盐晶种的球磨过程:将先前制备的金属碳酸盐粉末用陶瓷球在瓶中球磨3天。步骤(2):从瓶中收集经球磨的金属碳酸盐浆料,接着过筛。加上种晶技术的金属碳酸盐沉淀:步骤(3)至(5)步骤(3):金属硫酸盐的溶解过程:将硫酸镍六水合物、硫酸锰单水合物及硫酸钴七水合物溶于H2O中。此溶液的典型浓度为2mol/L。步骤(4):含有Na2CO3的金属碳酸盐前驱物的沉淀过程:金属碳酸盐沉淀的典型温度为90℃。在CSTR反应器中,搅拌速度为1000RPM。滞留时间为2小时。将金属碳酸盐晶种浆料一小时一次加至反应器中。步骤(5):金属碳酸盐前驱物的洗涤及干燥过程:将去离子水用于洗涤。所得湿饼在150℃下干燥超过16小时。种晶技术可高度控制金属碳酸盐沉淀期间的PSD,并且对于其他参数没有负面影响。首先,当在金属碳酸盐沉淀过程期间加入固定量但不同流速(CO3/M)的晶种(以金属碳酸盐浆料的形式)时,PSD受到种晶技术的控制及稳定化,而与不同流速(CO3/M)无关。然而,杂质含量仍与流速比强烈相关。其次,当在金属碳酸盐沉淀过程期间加入不同量的晶种并搭配固定流速(CO3/M)时,PSD会根据反应器中的晶种/产物比而改变,即使使用相同流速比。然而在此例中,杂质含量不受影响。这些实验结果显示于表5及6中。结论是在金属碳酸盐沉淀过程期间,PSD控制及稳定化通过种晶技术达成。由此可知,种晶技术能够在金属碳酸盐沉淀期间,视最终阴极产物的应用而调整粒径。还由此可知,一旦粒径得到控制,则流速比即决定Na/S比例。图9显示MCO3样本的PSD(D50,跨距)结果(■=不使用种晶的CO3/M系列的D50(μm),□=使用种晶的系列的D50(μm),●=不使用种晶的系列的跨距,o=使用种晶的系列的跨距),其来自相同晶种量及不同CO3/M比(参见表5)。图10显示MCO3样本的ICP(Na,S)结果(■=不使用种晶的CO3/M系列的Na(重量%),□=使用种晶的系列的Na(重量%),●=不使用种晶的系列的S(重量%),o=使用种晶的系列的S(重量%)),其来自相同晶种量及不同CO3/M比(参见表5)。最后,图11显示MCO3样本的敲紧密度(TD)结果(■=不使用种晶的CO3/M系列的TD(g/cm3),●=使用种晶的系列的TD(g/cm3)),其来自相同晶种量及不同CO3/M比(参见表5)。在各图中还给出不使用种晶的沉淀结果,其基于表1中的样本。表5:来自相同晶种量及不同CO3/M比的MCO3样本的分析结果表6:来自不同晶种量及固定CO3/M比1.08的MCO3样本分析结果实施例7:通过离子交换移除杂质除了PSD控制外,金属碳酸盐沉淀过程的另一个问题是杂质控制。为了降低硫含量,采用苛性洗涤,经洗涤的金属碳酸盐前驱物相较于金属氢氧化物前驱物具有相对低的硫含量,如在表7的苛性碱洗涤结果中所见。但是金属碳酸盐前驱物的钠含量比预期的高。应进行离子交换实验以研究化学品是否能够减少钠含量。因此,本实施例着重在具有高Na对S杂质比的前驱物,企图移除其中的杂质。这些前驱物以流速比CO3/M>1.00获得。此为量产所关注的议题,因为在这些环境中,所有过渡金属均会沉淀,而废水中残留的少量Na2CO3不是问题。通过控制离子交换实验中的洗涤时间、温度及添加剂种类,尝试减少钠含量。然而此实施例证明这过于困难,如果要有效减少钠杂质,需要过多时间或过于昂贵的化学品。表7:在苛性洗涤之后,来自CO3/M系列的MCO3样本的ICP(Na,S)结果表8:使用不同化学品进行离子交换实验所得的MCO3样本的ICP(Na,S)结果备注:LiOH*是指双重洗涤,MeSO4是指Ni0.6Mn0.2Co0.2SO4实施例8:使用含S及Na的MCO3前驱物制备及测试NMC阴极材料金属碳酸盐前驱物使用4L搅拌(1000rpm)反应器来制备。温度为90℃。将溶于水中的Na2CO3及MSO4的两个精确控制流连续注入反应器中。碱对酸流速比CO3/M为1.03。MSO4流的金属组成为M=NMC552。Na2CO3及MSO4流的浓度为2mol/L。滞留时间(即置换1整个反应器内容物所需的时间)为2.75小时。使用种晶技术。晶种通过球磨得自稍早沉淀的MCO3而获得。晶种的D50为0.5μm。将含晶种的浆料频繁注入反应器中,并且注入晶种及沉淀产物间的重量比为0.63%。沉淀在用水注满反应器一半之后开始。使沉淀执行12小时。从第4个小时开始收集溢流产物。在12小时之后将反应器内容物以及所收集的溢流重复过滤并在水中洗涤。以完全相同的方式重复进行沉淀数次以获得足够产物量。在沉淀期间每小时检测PSD。发现沉淀过程非常稳定,并且D50值的变化小于2μm,所得的D50为13±1.5μm。在过滤及洗涤之后,将产物在120℃下在空气中干燥过夜。将所获得的前驱物产物混合然后分析。敲紧密度为1.4g/cm3。ICP分析确认达到所期望的金属组成(552),该组成为Ni:Mn:Co=41.87:41.43:16.70。最终MCO3前驱物产物含有3300ppmNa及2400ppm硫,导致钠硫摩尔比为1.9,这在所期望的0.4<Na/S<2范围内。金属含量为49.9重量%,这稍高于无杂质MCO3的理论值(=48.80重量%),与SO4及Na杂质的存在相符。图12显示最终沉淀物的SEM剖面。粒子为相对致密并且没有观察到中空壳结构。许多粒子的形状接近球形。接下来,制备两个阴极粉末样本。一个样本基本上无杂质,另一个样本含有从MCO3前驱物产物残留下来的钠及硫杂质。无杂质样本(LX0142)的制备:将碳酸盐前驱物与Li2CO3共混,获得1.10的Li:M摩尔比(假设Li2CO3具有97%的纯度)。在10L/kg.min的空气流中,将2kg的此共混物缓慢加热至945℃,然后使烧结持续10小时。在冷却之后,于搅拌下将样本浸于水(1kg/2L)中10分钟、过滤然后干燥(在150℃下干燥16小时)。由于原始硫及钠杂质以可溶性Li2SO4、LiNaSO4或Na2SO4化合物存在,因此水处理可有效移除残余杂质。一般而言,水处理会化学损伤阴极材料粒子表面,导致实际电池中的循环稳定性不佳。因此采用“复原(healing)”热处理。(形态在水暴露期间不会实质改变)。在软式研磨之后,将已干燥的中间物样本在375℃下加热20小时。冷却之后将样本过筛。碳酸盐前驱物的粒径保持不变,所获得阴极的D50为14μm。含杂质样本(LX0143)的制备:将碳酸盐前驱物与Li2CO3共混,获得1.10的Li:M摩尔比(假设Li2CO3具有97%的纯度)。在10L/kg.min的空气流中,将2kg的此共混物缓慢加热至945℃,然后使烧结持续10小时。在冷却之后,将样本软式研磨,然后在375℃下再加热(类似于样本LX0143)20小时。(进行该再加热以利用如同样本LX0143的相同温度曲线来制备LX0142。我们预期未进行再加热的性能会相似)。在冷却后将样本过筛。优化阴极粉末参考物LX0031由致密的、10μm氢氧化物前驱物制备。条件类似于适合用于量产的条件。表9显示NMC阴极样本的ICP及表面积测量结果。图13显示下列阴极的SEM显微图:左侧:LX0031(由M(OH)2前驱物制备的参考物),中间:无杂质LX0142样本(中间物经洗涤),右侧:含杂质LX0143样本。图14显示样本LX0142的剖面SEM。显然可达到所期望的形态。粒子大致为球形并具有开放孔隙。在电池中,电解质将填满孔隙并且有利于Li快速扩散至粒子内部,从而实现高功率及低DCR。钮扣电池根据Umicore内部标准程序(RL4345N)来制备:电极制备如下:将约27.27重量%的活性阴极材料、1.52重量%聚偏二氟乙烯聚合物(KF聚合物L#9305,KurehaAmericaInc.)、1.52重量%导电碳黑(SuperP,ErachemComilogInc.)及69.70重量%N-甲基-2-吡咯啶酮(NMP)(来自Sigma-Aldrich)以高速均质机密切混合。接着将浆料在铝箔上以带铸法展布成薄层(典型100微米厚)。在蒸发掉NMP溶剂之后,使铸膜通过辊压(使用40微米间隙)处理。使用圆形冲模切刀(测得直径为14mm)从膜上冲压出电极。接着将电极在90℃下干燥过夜。之后将电极称重以确定活性材料含量。典型而言,电极含有90重量%的活性材料,活性材料装载重量为约17mg(~11mg/cm2)。接着将电极放在充氩手套箱中,并组装至钮扣电池体内。阳极为具有500微米厚度的锂箔(来源:Hosen);隔膜为Tonen20MMS微孔聚乙烯膜。钮扣电池填充有LiPF6溶于碳酸伸乙酯及碳酸二甲酯(1:2体积比)的混合物中的1M溶液(来源:TechnoSemichemCo.)。各电池在25℃下使用Toscat-3100计算机控制恒电流循环站(来自Toyo)来进行循环。测试规程如下:制备钮扣电池,其中电极由96重量%的活性材料组成。电极含量为约6mg/cm2。记录第一循环的放电容量(DQ1)、第一循环的不可逆容量(IRRQ1)及3C倍率相对于0.1C倍率的倍率能力(ratecapability,以%表示)。放电容量DQ1在25℃下,在4.3至3.0V范围的第一循环期间以0.1C测得(以mAh/g表示)。不可逆容量IRRQ1为(Q1C-DQ1)/Q1C(以%表示),Q1C为第一循环期间的充电容量。在Q0.1C及Q1C下的容量衰退以每100次循环的%表示。它们在快速1C倍率下检测25次循环期间的容量损失(比较循环7与34)以及在慢速0.1C倍率下检测25次循环期间的容量损失(比较循环8与35)并外插到100次循环而获得。Q1C(H)为在1C/1C循环下的衰退率,其通过比较得自循环36及60的22次循环期间的容量损失并将该损失外插到100次循环而获得。电化学测试结果示于表10中。表9:阴极的杂质ICP及表面积表10:阴极的钮扣电池测试结果结果证明1)得自含钠及硫碳酸盐(LX0142,LX0143)前驱物的NMC的表面积显著高于由密集M(OH)2前驱物制备的NMC参考物。SEM显微图明显指出大BET表面积源自开放孔隙,这通过剖面SEM确认。2)开放孔隙及高BET显著改善电化学性能。LX0142及143的不可逆容量显著低于参考物LX0031。针对给定的组成,充电容量基本上是固定值。因此如果不可逆容量减少,则可逆容量增加。所以相较于参考物具有低几乎2.5%(=9.94-12.44)不可逆容量的LX0143,产生相对增加的可逆容量(达3.2%)。3)含杂质样本LX0143的容量比无杂质样本LX0142低1.4%。这种较低容量与惰性碱硫酸盐的存在相符。钠及硫杂质以硫酸盐Li2NaSO4、LiNaSO4或Na2SO4存在,其不会增加可逆容量。我们估计有1.3重量%的盐存在。此完美解释了观察到样本LX0143的容量比LX0142低(-1.4%)。4)LX0142及LX0143的钮扣电池测试显示含杂质LX0143的循环稳定性比无杂质LX0142好。含有杂质的样本LX0143是本发明的一个实施例。无杂质LX0142通过更昂贵的制程所制备,因而不是工业上优选的。然而最重要的是,无杂质样本LX0142显示较低的循环稳定性。作者观察到,以所期望的比例及量存在的钠及硫杂质令人惊讶地造成改善的循环稳定性。实施例9:全电池测试制备全电池。全电池为卷袋(woundpouch)型电池并且具有约650mAh的容量。测试3种不同的电极材料:全电池批号AL705含有LX0031,其为得自氢氧化物的NMC参考物。全电池批号AL885含有无杂质LX0142而AL886含有含杂质阴极LX0143。整体而言,含有LX0143的AL886显示出优异结果(LX0143为具有所欲形态且含有在优选范围内的钠及硫杂质的NMC)。以下列示及论述电池制作及测试的细节。全电池组装为达全电池测试的目的,将所制备的正极(阴极)与负极(阳极,典型为石墨型碳)及多孔性电绝缘膜(隔膜)组装在一起。全电池通过下列主要步骤制备:(a)电极切割(electrodeslitting)(b)电极干燥(electrodedrying)(c)胶卷绕制(jellyrollwinding)(d)封装(packaging)。(a)电极切割:在NMP或水基涂布之后,电极活性材料可通过切割机来切割。电极宽度及长度根据电池应用来决定。(b)附接接头(tap):接头有两种。铝接头附接至正极(阴极),而铜接头附接至负极(阳极)。(c)电极干燥:在真空烘箱中将所制备的正极(阴极)及负极(阳极)在85℃至120℃下干燥8小时。(d)胶卷绕制:在电极干燥之后使用绕制机制造胶卷。胶卷是由至少一负极(阳极)、一多孔性电绝缘膜(隔膜)及一正极(阴极)所组成。(e)封装:用铝积层膜封装材将所制备的胶卷结合在800mAh电池中,形成袋式电池(pouchcell)。再来,将胶卷用电解质浸泡。电解质的量根据正极及负极、和多孔性隔膜的孔隙率及尺寸来计算得到。最后,将经封装全电池通过密封机密封。全电池评估许多不同的全电池评估测试皆为可能。本发明显示(a)循环稳定性、(b)容量及倍率能力、(c)膨胀、(d)储存测试及(e)DCR电阻测试的结果。(a)循环稳定性:电池经完全充电及放电数百次循环。循环测试在25℃或高温(例如45℃)下执行以加速非所期望的副反应,从而迫使更快速的容量损失。(b)容量及倍率能力:容量是以0.2C倍率的速度,在4.3V及2.7V之间测定的放电容量。效率第一次充电与第一次放电容量间的比例(以%表示)。倍率能力为以0.5、1.0、2.0、3.0及4.0C的倍率放电的容量,表示为在0.2C的倍率下的百分比。0.2C对应在5小时内将已充电电池放电的电流。举例来说,1C为比0.2C电流大5倍的电流。(c)膨胀:将袋式电池完全充电然后置于烘箱中,将其加热至90℃然后停留在此温度下数小时。在90℃下已充电电极会与电解质反应并产生气体。释出的气体会产生膨胀。在实施例中我们记述在高温暴露4小时之后所测得的厚度增加(=膨胀)值。膨胀对于许多应用而言是重要议题,此外作者预期膨胀是检测因涂布期间的水暴露所致的最终表面损伤的极敏感方法。(d)储存测试,即剩余及回复容量:将电池完全充电然后在60℃下储存1个月。在1个月之后将电池自60℃室取出,然后在25℃下测试。将电池放电,在放电期间测量剩余容量。在再充电之后,将电池放电,并获得回复容量。在此容量检验之后,将60℃下的储存再持续一个月,再度测量剩余及回复容量,接着将电池储存第三次,然后再次测量。除了与许多应用的相关性外,储存实验还为评估水基涂布期间阴极损伤的极敏感工具。(e)搭配储存测试进行的DCR电阻测试:在60℃储存1、2及3个月之后,除了测量容量外,还测量电池的DCR电阻及DCR随时间的变化(以相对于初始DCR的%表示)。DCR电阻得自对电流脉冲的电压响应,所使用的程序根据USABC标准(UnitedStatesAdvancedBatteryConsortiumLLC)。DCR电阻与实际应用极为相关,因为数据可用来将衰退率外插到未来以预测电池寿命,此外DCR电阻对于检测电极损伤极为敏感,因为电解质与阳极或阴极间的反应的反应产物会沉淀为低导电度的表面层。表11显示全电池的容量及倍率能力结果。倍率能力(以相对于C-倍率的%表示)还显示在图15中。表12显示膨胀测试结果及全电池温度测试的温度特性。表13显示全电池测试的高温储存结果(DCR及DCR增加测试结果)。图16显示全电池在室温下的循环稳定性结果,而图17显示45℃下的这些结果。一般而言,制造自金属碳酸盐前驱物的锂金属氧化物产物比起制造自金属氢氧化物前驱物者,在全电池中具有优选的电化学特性:首先,制造自金属碳酸盐前驱物的锂金属氧化物产物具有较高的放电容量及较高效率。其次,这些产物具有较低DCR(在低SOC下)及较高倍率能力。应注意的是,这些电化学特性在电动车应用中是重要参数。在比较无杂质LX0142与LX0143的结果时,我们观察到类似于钮扣电池测试的结果,即含杂质样本LX0143有明显优选的优异循环稳定性。作者将改善的循环稳定性归因于所欲量的钠及硫杂质的存在。表11:全电池测试的容量及倍率能力表12:膨胀测试结果及全电池温度测试的温度特性表13:全电池测试的高温储存结果实施例10:来自含S及Na的MCO3的NMC中的杂质当含钠及硫MCO3用于制备NMC阴极时,大部分的钠及硫以杂质形式留在最终样本中。如果钠硫杂质比低于1,我们预期钠杂质以Li2SO4及LiNaSO4存在。如果钠硫杂质比介于1和2之间,我们预期Na2SO4及LiNaSO4会共同存在。LiNaSO4可在结晶学数据库中在ICSD编号#3814下找到。主峰在约2θ=23°。为了确认这点,LiNaSO4通过使Na2S2O8与Li2CO3在400℃下反应来制备。反应方程式为Li2CO3+Na2S2O8→2LiNaSO4+CO2+1/2O2。基本上会获得单相LiNaSO4。图18显示XRD图谱以及使用来自ICDS数据库针对#3814的晶格常数及原子位置所计算得到的图谱。LiNaSO4相的主峰在2θ的23.44°。其他强峰在32.74°及22.66°。尤其是如果钠硫比接近1(在此情况中大部分杂质相为LiNaSO4),杂质相的X射线散射可能强到足以由粉末XRD所清楚检测。另一方面,在23.44、32.74及22.66度检测到不同峰为LiNaSO4相存在的强力证据。实施例11:由含钠及硫MCO3前驱物制备富含Li及锰的阴极具有NMC=261组成的实验工厂前驱物使用种晶技术来获得,如实施例1及6中所解释的。图19显示该前驱物的SEM显微图。该前驱物含有3216ppm钠及5280ppm硫(通过ICP所测得)。将该前驱物与Li2CO3共混。Li:M共混比为1.468。制备1kg。将共混物缓慢加热至800℃,接着烧制10小时。在冷却之后将产物过筛,得到样本HLM330。最终产物的形态示于图20中。达成所欲形态。粒子为球状且相对致密但同时又展现显著孔隙。BET表面积相对为大:4.8m2/g,远大于相同形状的致密粉末,代表开放中孔结构存在。在烧制之后,预期总共2重量%杂质,其中86%(17625ppm)的杂质以LiNaSO4,而14%以Li2SO4存在。一般而言,如果钠硫比接近一,我们预期最高贡献来自LiNaSO4。该材料在钮扣电池中具有优异性能(如实施例8中所述制备)。当在室温(25℃)下使用80mA/g的电流在3.0至4.6V之间进行测试时,会达到291mAh/g的可逆容量及5.4%的不可逆容量。这些是杰出的成果,这显著高于由M(OH)2制备的类似材料所能达到的任何数据。循环稳定性令人满意:每100次循环衰退16%。进行仔细的慢速XRD研究。事实上,可清楚检测到作为次要相的LiNaSO4相。使用0.02的步宽扫描15至40°的范围4小时(0.1°/min)。图21及22显示所得XRD图谱(a.u.对度°)。在图21中,y轴为对数尺度,所以杂质峰得到强化。在23.2、29.54、30.32及32.55°的峰明显可归因于XRD图谱。在图22中,y轴为线性尺寸。展开图放大总量值0.5至5%的区域。在23.2、29.54、30.32及32.55°的峰明显可归因于LiNaSO4次要相。实施例12:来自含S及Na的MCO3的532NMCMCO3前驱物通过如实施例1中所述的类似方法来制备,除了所沉淀出的碳酸盐由具有NMC=552改为具有NMC=532组成。晶种从球磨得自先前沉淀的MCO3而获得。碱/酸(CO3/M)流比设定在1.03。所沉淀的碳酸盐(样本MCO-0099ak)具有D50=16μm。观察到具有显著纳米孔隙(BET=144m2/g)的良好球形形态。图23显示所沉淀碳酸盐的SEM。然而,ICP显示钠硫杂质比不在所期望范围0.4<Na/S<2内。杂质含量为5370ppmNa及2400ppm硫。这导致钠硫杂质比>3。将前驱物MCO-0099ak与Li2CO3共混然后烧制。用不同Li:M比及不同烧制温度来制备数个最终样本。样本通过钮扣电池测试来测试,如实施例8。尽管有优异形态、高表面积及正确晶体结构(微晶尺寸及晶格常数),一般而言观察到相当令人失望的电化学特性。给出样本EX1518及EX519的典型结果。这些样本具有与由M(OH)2前驱物制备的NMC532类似的性能。针对样本EX1518及EX519,Li:M共混比为1.02且烧结温度分别为900℃(EX1518)及875℃(EX1519)。表14汇总了所获得的结果。样本60893是由NMC532金属氢氧化物制备的参考样本。可逆容量在25℃下,以16mA/g在3.0至4.3V之间测量。我们假设Na含量过高且硫含量过低是性能不佳的原因。因此将附加Li2SO4加至共混物。在过量硫存在下,非期望的钠将会从晶体结构中移除。简化的反应方程式为LiM1-xNaxO2+xLi2SO4→Li1+xM1-xO2+xLiNaSO4重复样本制备。将MCO3前驱物MCO-0099ak与Li2CO3共混。Li:M共混比为1.02。然而在此将每1过渡金属2mol%Li2SO4加至共混物。在加入硫酸盐之后,共混物中的钠硫杂质比即在所期望范围0.4至2.0内。将共混物在875℃下烧制10小时,得到样本EX1534。图24显示该样本的SEM。图25显示样本EX1534的X射线粉末绕射图谱。扫描条件为2小时扫描、0.02°步宽、15至85°。LiNaSO4次要相明显存在,因为在22.58、23.22、29.53、30.39及32.53°观察到峰。y轴为对数尺度以强化小型峰。钮扣电池测试显示电化学性能有显著改善(参见表14)。尽管加入电化学“惰性”硫酸盐,可逆容量仍增加。该增加是由于不可逆容量从约11大幅减少至8%所致。为了移除Na及硫杂质,将样本EX1534的其余部分用水洗涤以移除可溶性Li及Na硫酸盐。在过滤之后,将样本在700℃下热处理5小时。将所得样本EX1535在钮扣电池中进行测试。达到优异容量结果。不可逆容量进一步减少到7%而可逆容量则达到176mAh/g,此对于NMC532而言为相当高的值。此极高值(相较于EX1534)由于(1)极低不可逆容量及(2)移除电化学惰性硫酸盐所致。此外,大BET表面积(2.14m2/g)促成了大可逆容量。然而无杂质样本显示在全电池中较差的循环稳定性(数据未显示)。表14:NMC532的性能表14:接续实施例13:来自含S及Na的MCO3的532NMC在实施例12中,样本EX1518及EX1519获得的结果不佳。为了进一步研究此性能是否是由过高钠硫杂质比所造成,选用具有所期望范围内的较低比例的前驱物,然后重复实验。所选用的MCO3前驱物通过类似于实施例1中所述的方法来制备:1)将碱对酸流速比(CO3/M)调整至1.00以在所期望0.4<Na/S<2范围内达成良好的钠及硫杂质平衡。2)未应用种晶3)沉淀执行6小时,样本从第4小时开始收集所得样本MCO-0112a含有2230ppm钠及4190ppm硫,并且钠硫杂质比为0.74。杂质在所期望的杂质范围内。将前驱物与Li2CO3混合。Li:M共混比为1.02。将共混物在875℃下的空气流中烧制10小时,得到样本EX1577。大部分Na及S杂质残留并且ICP分析显示最终NMC含有2498ppmNa及4372ppm硫。EX1577具有2.12m2/g的高BET表面积、优选的球形、中孔粒子形态(如在图26(SEM显微图)中所示)及优异电化学性能。达到6.1%的极低不可逆容量,可逆容量为174.9mAh/g(使用一般条件:3.0至4.3V,16mA/g,25℃)。来自实施例12的样本EX1518及EX1519具有类似形态,并且晶体结构的细节(晶格体积、结晶度)还极为相似。然而,观察到比EX1577要差的电化学性能。作者相信EX1518及EX1519性能不佳的主要原因是碳酸盐前驱物中的高钠硫杂质比,其超过所期望的0.4<(Na/S)<2范围。相反地,样本1577由该比在优选范围内的前驱物制备。实施例14:来自含S及Na的MCO3的111NMC具有NMC=111组成的含钠及硫碳酸盐前驱物如实施例1中所述制备。碱/酸流比(CO3/M)选择为1.0以达到在所期望范围0.4<Na/S<2内的钠硫杂质比。所得样本MCO-0114g含有2890ppm钠及3660ppm硫,因此钠硫杂质比为1.1。将前驱物与Li2CO3混合。Li:M共混比为1.1。将共混物在850℃下的空气流中烧制10小时,得到样本MX0809。MX0809具有高BET表面积、优选的球形、中孔粒子形态(参见图27的SEM显微图)及优异电化学性能。达到4.4%的极低不可逆容量,可逆容量为161.3mAh/g(使用一般条件:3.0至4.3V,16mA/g,25℃)。由致密M(OH)2制备的典型量产参考物具有>10%的不可逆容量,且可逆容量为约155mAh/g。作者将此优异电化学性能归因于(a)使用MCO3基前驱物而来的开放中孔,以及(b)MCO3前驱物具有优选杂质比范围内的钠及硫杂质实施例15:使用干粉进料的含Na及硫碳酸盐的沉淀先前实施例已显示具有优异电化学性能的NMC可自含硫及钠碳酸盐前驱物达成。然而,相较于量产的典型M(OH)2沉淀,MCO3沉淀的主要缺点在于其体积效率较低。主要原因是Na2CO3的溶解度远低于NaOH。在典型M(OH)2沉淀中,进料可为(1)10MNaOH、(2)2MMSO4及(3)10MNH4OH。沉淀根据(经简化的)下式:2NaOH+MSO4+NH4OH→Na2SO4+M(OH)2+NH4OH这显示要沉淀1mol的M(OH)2,需要200mlNaOH、500mlMSO4及100mlNH4OH。这加起来等于每1mol沉淀过渡金属氢氧化物需要800ml溶液。假使NMC=532,则1mol的过渡金属氢氧化物M(OH)2对应于91.6克。以典型MCO3沉淀而言,进料可为(1)2MNa2CO3及(2)2MMSO4,其中两个流均具有与溶解度上限相差不远的浓度。沉淀根据(经简化的)下式:Na2CO3+MSO4→MCO3+Na2SO4。这显示要沉淀1mol的MCO,需要500ml的Na2CO3及500ml的MSO4。这加起来等于每1mol沉淀MCO3需要1L。假使NMC=532,则1mol的沉淀金属碳酸盐MCO3对应于117.6克。就效率的观点而言,碳酸盐沉淀必须处理更大量(+25%)的废水。固体处理(过滤、干燥等)也会导致与沉淀物的体积或质量成正比的成本。由于过渡金属含量在MCO3中比M(OH)2要低(即49%对62%),因此对于碳酸盐沉淀而言固体处理的效率倾向较低。最后,NaOH可用具竞争力的价格购得为液体,而Na2CO3以粉末形式购得并且需要现地溶解的设备。因为以上所有原因,MCO3沉淀可能效率较低,因此对于提高碳酸盐沉淀的效率有强烈需求,以达成本方面真正具有竞争力的制程。此实施例建议一种沉淀途径,其中基本上连续地将MSO4流及同时地Na2CO3粉末馈入搅拌反应器中。固体馈入可通过重力控制的螺杆进料机来进行。此方法会将每mol沉淀MCO3的1L废水减少50%至500ml。至于在实验室进行的实验,因为没有重力控制的螺杆进料机可用,因此每10分钟加入固定量的Na2CO3粉末,而MSO4则连续馈入。除了以粉末取代Na2CO3流之外,实验类似于实施例1所述。金属组成为NMC552。提高MSO4流速以达到约2.5小时的相同滞留时间。碱对酸比(CO3/M)在不同时间下均固定在1.03。此沉淀会达成钠硫杂质比在优选范围0.4<Na/S<2内的MCO3。所沉淀的MCO3含有2649ppmNa及8086ppm硫(Na/S=0.45)。总杂质浓度高于液体沉淀所预期的。作者相信较高杂质水平由于制程控制不佳所致。得到较少杂质的优选制程可通过精确及连续馈入Na2CO3粉末来达成。相较于正常沉淀,该沉淀碳酸盐较不致密且较为蓬松。优选制程控制应能同时显著改善这些问题。图28示意说明为什么非连续固体馈入可造成较高的总杂质,以及为什么较为连续的制程可解决此问题。线B-1-A为采用固定流速的“正常”沉淀所获得的杂质线(参见实施例1-3)。当CO3/M提高时,Na增加且硫减少,但该线不为直线。点(1)显示在正常沉淀之后所获得的杂质,例如在1.0的流速比(CO3/M)下。在加入Na2CO3粉末之后,CO3/M比暂时提高且具有高Na杂质(点“A”)的MCO3沉淀。随着Na2CO3消耗(未加入Na2CO3一段时间但连续注入MSO4溶液),溶液中的CO3/M比降低。在点“B”,富含硫的MCO3沉淀。最终MCO3产物将会是具有不同杂质比的MCO3混合物。然而,碳酸盐“A”及“B”的混合物为点2。点2的总杂质含量高于在点“1”所沉淀的参考氢氧化物。作者预期,当Na2CO3粉末的加入变成连续,点“A”及“B”均朝向点“1”移动并且可达成优选的MCO3产物。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1