用汉逊酵母表达系统产生HPV6L1蛋白的方法与流程
用汉逊酵母表达系统产生HPV6L1蛋白的方法发明领域本发明属于医药生物工程技术领域,涉及产生HPV6L1蛋白的方法,尤其涉及用汉逊酵母表达系统产生HPV6L1蛋白的方法。
背景技术:
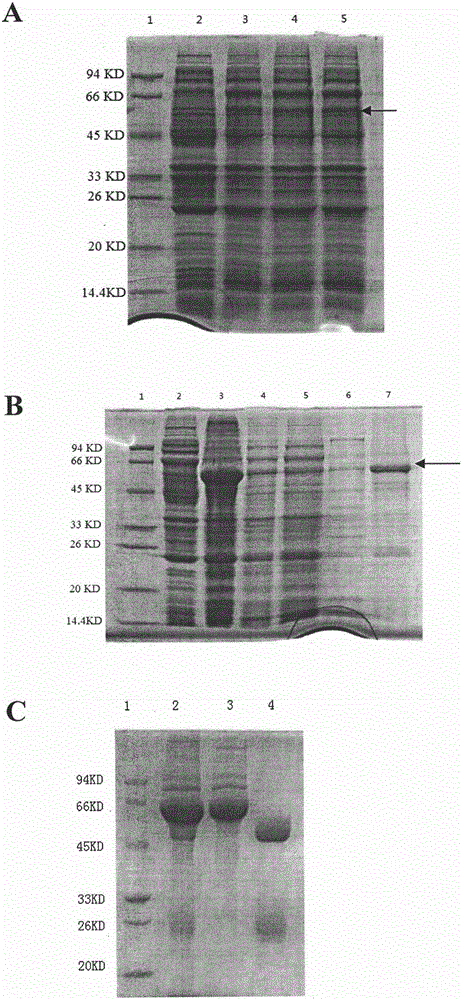
人乳头瘤病毒(humanpapillomavirus,HPV)是一种无包膜的闭环双链DNA病毒,属乳多空病毒科多瘤病毒亚科,主要侵犯人体的上皮黏膜组织,进而诱发各种良性及恶性增生病变。目前已鉴定出来的不同亚型HPV超过200种,HPV感染具有明显的组织特异性,不同型别的HPV对于皮肤和黏膜的嗜向性不同,能诱发不同的乳头状病变,大约有30多种HPV型别与生殖道感染有关,其中有20多种与肿瘤相关。根据HPV诱发病变的良恶性不同,HPV可大致分为两类:1)高危型(如HPV16、HPV18、HPV45、HPV31、HPV33、HPV52、HPV58、HPV35、HPV59、HPV56、HPV39、HPV51等):与人类多种组织恶性肿瘤密切相关,主要引起重度不典型增生和浸润癌,尤其以HPV16及HPV18型感染与宫颈癌的发生最为密切,这两个型别感染占宫颈癌感染的70%以上,其中HPV16占50%以上;2)低危型(如HPV6、HPV11、HPV40、HPV42、HPV43、HPV44、HPV54、HPV72、HPV81等):可引起表皮细胞良性增殖性性病,如尖锐湿疣和扁平湿疣等,其中由HPV6与HPV11诱发的尖锐湿疣占90%以上。HPV主要由病毒外壳和基因组DNA构成。基因组长约7900bp,有8个病毒蛋白编码基因。其中6个ORF编码的蛋白在病毒复制的早期表达,称为早期蛋白;2个ORF编码的蛋白在病毒复制的晚期表达,称为晚期蛋白。晚期蛋白包括主要外壳蛋白L1及次要外壳蛋白L2,并参与病毒外壳的形成。HPV病毒外壳蛋白能够进行自组装,在多种表达系统中单独表达的L1蛋白或将L1蛋白与L2蛋白共表达时均能自组装成病毒样颗粒(virus-likeparticle,VLP),其中以在酵母系统、杆状病毒昆虫表达系统及哺乳动物细胞表达系统等产生的VLP更接近天然病毒的结构。利用外源表达体系生产的VLP免疫后能够在体内诱发产生中和抗体,获得良好的免疫保护效果。多形汉逊酵母(HansenulaPolymorpha),又称作Pichiaaugusta,是当前公认的最为理想的外源基因表达系统之一。多形汉逊酵母作为单细胞真核微生物,既具备原核生物生长快速、易于遗传操作等特点,又有真核细胞翻译后加工和修饰等功能。此外,汉逊酵母还具备安全性好、易于培养、成本低廉、表达量高及遗传稳定等优势,并且能够克服诸如酿酒酵母(Saccharomycescerevisiae)菌株不稳定、产量低及糖基化侧链过长以及毕赤酵母(PichiaPastoris)外源基因整合拷贝数较低的问题。目前,应用汉逊酵母表达系统生产的药物(如胰岛素,商品名Wosulin)及HBV疫苗(商品名Hepavax-Gene)均已上市销售。发明概述在第一个方面,本发明提供了一种产生表达HPV6L1蛋白的重组汉逊酵母细胞的方法,其包含以下步骤:a)通过将包含编码HPV6L1蛋白的核苷酸序列的外源多核苷酸插入载体来构建表达构建体;b)用步骤a)中获得的表达构建体转化汉逊酵母细胞;和c)对步骤b)中获得的汉逊酵母细胞进行筛选,获得含有所述外源多核苷酸的重组汉逊酵母细胞。在第二个方面,本发明还提供了根据上述方法产生的重组汉逊酵母细胞。在第三个方面,本发明还提供了一种产生HPV6L1蛋白的方法,包括以下步骤:i)在适于HPV6L1蛋白表达的条件下培养本发明的重组汉逊酵母细胞;和ii)从培养物中回收并纯化HPV6L1蛋白。所述方法还可以包括对所纯化的HPV6L1蛋白进行解聚和重聚的步骤。在最后一个方面,本发明还提供了根据本发明的方法产生的HPV6L1蛋白在制备用于预防HPV6感染的疫苗中的用途。附图说明图1以MOX启动子片段为探针的Southern印迹检测结果。以ATCC26012基因组DNA为对照。1:对照(上样量为:1000ng);2:对照(上样量为:500ng);3:对照(上样量为:250ng);4:对照(上样量为:125ng);5:HP/pRMHP2.1-6wt基因组DNA(上样量为:15.625ng,其亮度介于500ng和1000ng对照之间);6:HP/pRMHP2.1-6sc基因组DNA(上样量为:15.625ng,其亮度介于500ng和1000ng对照之间);7:HP/pRMHP2.1-6hp基因组DNA(上样量为:15.625ng,其亮度介于500ng和1000ng对照之间)。图2AHPV6L1蛋白在大肠杆菌中诱导表达的SDS-PAGE结果。1:蛋白标志物;2:未诱导的对照样品;3,4,5:IPTG诱导表达样品。图2B原核表达的HPV6L1蛋白的纯化结果。1:蛋白标志物;2:超声破碎后的上清液;3:超声破碎后的沉淀部分;4,5:流穿液;6:20%洗脱液;7:100%洗脱液。图2C兔多克隆抗体纯化结果。1:蛋白标志物;2:抗血清;3:流穿液;4:洗脱抗体。图3不同的重组汉逊酵母细胞表达水平的Western印迹检测。1:HP/pRMHP2.1-6wt;2:HP/pRMHP2.1-6sc;3,4:HP/pRMHP2.1-6hp;5:标准品(30μg/L);6:预染蛋白标志物。图4发酵过程中HPV6L1蛋白的表达检测。1:预染蛋白标志物;2:标准品(30μg/L);3:诱导0小时;4:诱导3小时;5:诱导6小时;6:诱导9小时;7:诱导10小时。图5AHPV6L1蛋白的POROS50HS纯化电泳图。1:上柱样品;2:流穿液;3~9:NaCl梯度洗脱。图5BHPV6L1蛋白的CHT纯化电泳图。1:上柱样品;2∶3~6∶磷酸盐梯度洗脱。图6纯化的HPV6L1蛋白的透射电镜观察结果。发明详述本发明人成功地建立了利用汉逊酵母产生HPV6L1蛋白的方法,所产生的HPV6L1蛋白能够自组装成病毒样颗粒,可用于制备预防HPV感染的疫苗。本发明首先提供了一种产生表达HPV6L1蛋白的重组汉逊酵母细胞的方法,其包含以下步骤:a)通过将包含编码HPV6L1蛋白的核苷酸序列的外源多核苷酸插入载体来构建表达构建体;b)用步骤a)中获得的表达构建体转化汉逊酵母细胞;和c)对步骤b)中获得的汉逊酵母细胞进行筛选,获得含有所述外源多核苷酸的重组汉逊酵母细胞。本发明还包括根据所述方法产生的重组汉逊酵母细胞。来源于不同HPV6病毒株的HPV6L1蛋白的氨基酸序列会存在差异。本发明人通过对数据库中所有的HPV6L1蛋白序列进行比对,在HPV6L1蛋白每个氨基酸位置上选取出现频率最高的氨基酸残基,获得了示于SEQIDNO:1的氨基酸序列,该序列是HPV6L1蛋白最具代表性的共有序列。因此,本发明中的HPV6L1蛋白优选具有SEQIDNO:1所示的氨基酸序列。为了利用汉逊酵母高效地表达HPV6L1蛋白,发明人根据SEQIDNO:1所示的氨基酸序列,针对汉逊酵母进行核苷酸序列的密码子优化。优化原则包括:a)按照汉逊酵母遗传密码使用频率表(http://www.kazusa.or.jp/codon/cgi-bin/showcodon.cgi?species=4905)选用使用频率最高或较高的密码子;b)避免对基因转录或蛋白翻译有潜在影响的负调控元件,如PolyAT区、PolyGC区、沉寂子(Sliencer)区及内部剪接位点等;c)对包含5’端UTR、HPV6L1编码区及3’端UTR在内的mRNA二级结构进行综合分析,避免复杂RNA二级结构的形成,使mRNA二级结构的自由能降低;d)在编码区上游尽可能采用与汉逊酵母启动子下游天然序列完全一致的5’UTR区;e)消除常用的限制性内切酶识别位点。经过优化的核苷酸序列示于SEQIDNO:4。本发明中所使用的编码HPV6L1蛋白的核苷酸序列优选为SEQIDNO:4所示的序列。本发明可以应用的汉逊酵母表达载体为申请号为201210021524.X的中国专利申请中描述的汉逊酵母表达载体pRMHP2.1(包含示于SEQIDNO:15的序列)。通过将包含编码HPV6L1蛋白的核苷酸序列的外源多核苷酸克隆进pRMHP2.1载体,可以获得本发明的表达构建体。本领域技术人员应当了解,本发明的表达构建体还可以用其它载体构建,例如已授权的中国专利CN100400665C中所描述的载体。为了能在汉逊酵母中表达,所述表达构建体中的外源多核苷酸可操纵地与启动子和终止子连接。如本文所用,“可操纵地连接”指至少两个多核苷酸的功能连接。例如,可操纵地连接包括启动子与另一个多核苷酸之间的连接,其中所述启动子序列起始并介导该另一个多核苷酸的转录。可操纵地连接包括终止子与另一个多核苷酸之间的连接,其中所述终止子终止该另一个多核苷酸的转录。适用于本发明的启动子包括但不限于MOX、FMD、AOX1和DHAS启动子。在一些实施方案中,本发明中使用的启动子是来自汉逊酵母的MOX启动子。在另一些实施方案中,本发明中使用的启动子是来自汉逊酵母的FMD启动子。适用于本发明的终止子包括但不限于来自汉逊酵母的MOX终止子。将表达构建体转化至汉逊酵母细胞可以用多种本领域已知的方法进行,包括但不限于电穿孔和PEG介导的转化。另外,本领域中已经开发多种用于表达外源蛋白汉逊酵母菌株,包括但不限于CGMCC2.2498汉逊酵母、ATCC34438汉逊酵母和ATCC26012汉逊酵母细胞。这些汉逊酵母菌株也可以应用于本发明。在一些实施方案中,用于转化本发明的表达构建体的汉逊酵母细胞是ATCC26012汉逊酵母细胞。在转化后,可以依据载体上携带的抗性基因来选择重组汉逊酵母细胞。合适的抗性基因包括但不限于Kan抗性基因和Zeocin抗性基因。依据所使用的载体,也可能使用营养缺陷培养基来筛选重组汉逊酵母细胞。外源多核苷酸在汉逊酵母中的表达水平与其在汉逊酵母中的拷贝数正相关。因此,还可以通过Southern印迹或定量PCR等方法进行进一步的筛选含有多拷贝外源多核苷酸的重组汉逊酵母细胞。优选外源多核苷酸拷贝数大于5的重组汉逊酵母细胞,更优选外源多核苷酸拷贝数大于30的重组汉逊酵母细胞。在另一方面,本发明还提供了一种产生HPV6L1蛋白的方法,包括以下步骤:i)在适于HPV6L1蛋白表达的条件下培养本发明的重组汉逊酵母细胞;和ii)从培养物中回收并纯化HPV6L1蛋白。本领域已知可用于培养汉逊酵母的各种培养基和基本培养条件,本领域技术人员能够根据需要进行选择或修改。本发明的重组汉逊酵母表达菌株的培养根据所需蛋白量可以在烧瓶进行或在不同规模的生物反应器(如30L的发酵罐)进行。根据所选用的启动子,可以在培养中加入合适的诱导物来诱导所述HPV6L1蛋白的表达。在使用MOX或FMD启动子的情况下,加入甲醇作为诱导物。所产生的HPV6L1蛋白的纯化可以使用本领域已知的各种蛋白纯化方式,如盐析、超滤、沉淀、层析等或这些方式的组合。在一个实施方案中,首先用层析介质POROS50HS(AppliedBiosystems)进行初步纯化,随后利用层析介质Macro-Prep陶瓷羟基磷灰石(TypeII,40μm)进一步纯化。利用本发明的方法制备的纯化的HPV6L1蛋白可以自组装成病毒样颗粒(实施例9,图6),并在小鼠中显示出良好的免疫原性(实施例10),因此本发明也提供了所述HPV6L1蛋白在制备用于预防HPV6感染的疫苗中的用途。实施例下面将通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所描述的实施例范围中。实施例1:HPV6L1共有氨基酸序列的分析全长的HPV6L1蛋白由500个氨基酸组成,经过GenBank检索,共获得含有500个氨基酸的全长HPV6L1蛋白序列117条。使用VectorNTI软件AlignX功能进行氨基酸序列比对分析,获得最具代表性的HPV6L1共有氨基酸序列(consensusaminoacidsequence,即在HPV6L1每个氨基酸位置均采用出现概率最高的氨基酸残基的序列),其序列如SEQIDNO:1所示。实施例2:HPV6L1编码基因的人工合成本发明共合成了3种不同的HPV6L1核苷酸序列,分别称为6wt、6sc和6hp:1)6wt-与GenBank登录号AF335604.1所示的天然HPV6L1基因序列相同的核苷酸序列,示于SEQIDNO:2;2)6sc-本发明人根据SEQIDNO:1所示的氨基酸序列,针对酿酒酵母遗传密码偏爱性全新设计的核苷酸序列,序列示于SEQIDNO:3;3)6hp-本发明人根据SEQIDNO:1所示的氨基酸序列,针对汉逊酵母遗传密码偏爱性全新设计的核苷酸序列,序列示于SEQIDNO:4。根据以上核苷酸序列,委托北京诺赛基因组研究中心有限公司分别进行6wt、6sc及6hp的全序列人工合成,克隆于T载体中(分别命名为T-6wt、T-6sc及T-6hp),并对其进行测序验证。实施例3:产生携带不同HPV6L1核苷酸序列的表达构建体本发明所应用的汉逊酵母表达载体为申请号为201210021524.X的中国专利申请中描述的汉逊酵母表达载体pRMHP2.1(其包含SEQIDNO:15所示序列)。(1)MOX启动子及MOX终止子的PCR扩增以汉逊酵母菌株ATCC26012及ATCC34438的混合基因组DNA为模板,用以下引物对获得大小为1518bp的MOX启动子,同时在上游引入NotI酶切位点;MOX启动子引物:5’-AAGGAAAAAAGCGGCCGCAACGATCTCCTCGAGCTGCTCGC-3’(SEQIDNO:16)MOX启动子引物:5’-TTTGTTTTTGTACTTTAGATTGATGTC-3’(SEQIDNO:17)以汉逊酵母菌株ATCC26012及ATCC34438的混合基因组DNA为模板,用以下引物对获得大小为311bp的MOX终止子,同时在下游引入BglII酶切位点;MOX终止子引物:5’-GGAGACGTGGAAGGACATACCGC-3’(SEQIDNO:18)MOX终止子引物:5’-GAagatctCAATCTCCGGAATGGTGATCTG-3’(SEQIDNO:19)(2)产生携带不同HPV6L1核苷酸序列的表达构建体以携带6wt的重组质粒T-6wt为模板,以引物1与引物2扩增获得大小为1551bp的HPV6L1wt基因,同时在上游引入MOX启动子区3’端的重叠序列,在下游引入MOX终止子5’端的重叠区;引物1:5’-CATCAATCTAAAGTACAAAAACAAAATGTGGCGGCCTAGCGACAGCACAG-3’(SEQIDNO:5)引物2:5’-GCGGTATGTCCTTCCACGTCTCCTTACCTTTTAGTTTTGGCGCGCTT-3’(SEQIDNO:6)以携带6sc的重组质粒T-6sc为模板,以引物3与引物4扩增获得大小为1551bp的HPV6L1sc基因,同时在上游引入MOX启动子区3’端的重叠序列,在下游引入MOX终止子5’端的重叠区;引物3:5’-CATCAATCTAAAGTACAAAAACAAAATGTGGAGACCATCTGATTCTACTG-3’(SEQIDNO:7)引物4:5’-GCGGTATGTCCTTCCACGTCTCCTTATCTTTTCGTCTTGGCTCTTTTC-3’(SEQIDNO:8)以携带6hp的重组质粒T-6hp为模板,以引物5与引物6扩增获得大小为1551bp的HPV6L1hp基因,同时在上游引入MOX启动子区3’端的重叠序列,在下游引入MOX终止子5’端的重叠区;引物5:5’-CATCAATCTAAAGTACAAAAACAAAATGTGGAGACCATCGGACTCTACTG-3’(SEQIDNO:9)引物6:5’-GCGGTATGTCCTTCCACGTCTCCTTAGCGCTTCGTCTTCGCTCTTTTC-3’(SEQIDNO:10)分别以MOX启动子、6wt基因/6sc基因/6hp基因、MOX终止子三个片段为模板,以引物7与引物8扩增获得大小为3.4Kb的6wt表达盒/6sc表达盒/6hp表达盒,同时在上游携带NotI酶切位点,在下游携带BglII酶切位点。引物7:5’-AAGGAAAAAAGCGGCCGCAACGATCTCCTCGAGCTGCTCGC-3’(SEQIDNO:11)引物8:5’-GAagatctCAATCTCCGGAATGGTGATCTG-3’(SEQIDNO:12)通过NotI+BglII双酶切分别将6wt表达盒、6sc表达盒及6hp表达盒克隆入pRMHP2.1载体中,获得表达构建体pRMHP2.1-6wt、pRMHP2.1-6sc及pRMHP2.1-6hp。实施例4:重组汉逊酵母细胞的产生(1)重组表达构建体质粒的提取及酶切挑取转化入实施例3中获得的重组表达构建体质粒的大肠杆菌菌落,扩大培养后使用E.Z.N.APlasmidMiniKit试剂盒(OmegaBio-Tek公司)提取质粒,并用BglII进行单酶切,应用E.Z.N.AGelExtractionKit试剂盒(OmegaBio-Tek公司)进行回收,用60μL预热至55℃的无菌水进行洗脱,通过测定OD260进行DNA定量,并将线性化的片段稀释至100nng/μl,保存于-20℃冰箱中,备用。(2)汉逊酵母细胞的处理挑取汉逊酵母菌株ATCC26012单菌落,接入含5ml的YPD液体培养基的小试管中,37℃培养12小时;取菌液5ml转接至200mlYPD培养基中,37℃培养4-6小时,至OD600约为1.0-1.5,于6000rpm离心10min;用200ml0.1mol/L磷酸盐缓冲液(含25mmol/LDTT,pH7.5)重悬菌体,充分混匀,于37℃孵育30min,6000rpm离心10min,弃上清,留菌体。用预冷的STM溶液200ml洗菌体,菌体吹吸均匀,于4℃6000rpm离心3min,弃上清,留沉淀。用冰冷的STM溶液100ml重悬菌体,于4℃6000rpm离心3min,弃上清,留沉淀。根据菌体量用100-200μl冰冷的STM溶液重悬菌体,并将菌液转移至高压后的离心管中,冰浴,准备转化。(3)汉逊酵母细胞的电转化按质粒∶菌体=1∶2的量加入重组汉逊酵母表达质粒15μl,菌液30μl,充分吹吸均匀,置于冰浴中待转化;将事先用酒精浸泡,且紫外照射后,于-20℃冷藏的电转杯取出,加入质粒菌体混合液;按电压2500V、电阻150Ω、电容50μF的条件进行电击;电击后迅速加入1ml已平衡至室温的YPD溶液,轻轻混匀后转移至EP管中;将电转后的菌体于37℃水浴中放置1h,每间隔15min轻轻颠倒3次;将已孵育1h的菌液,于6000rpm离心10min,弃上清;用200μlYPD溶液重悬菌体,以100μl/板涂布于含0.25mg/mlZeocin的YPD平板中,于37℃倒置培养3-7天。(4)重组汉逊酵母表达菌株的传代、稳定挑取Zeocin抗性平板上长出的重组菌株单菌落,接种于5ml含0.25mg/mlZeocin的YPD液体培养基中,于37℃、200rpm摇床培养24-36小时,至OD值达50后,以1∶1000的比例转接于5ml含0.25mg/mlZeocin的YPD液体培养基中,培养至OD值达50后,再以1∶1000的比例转接于5ml含0.25mg/mlZeocin的YPD液体培养基中,以此类推,连续传10次并进行菌种的保存。保存体系为菌液∶60%甘油=1∶1,根据需要量保存菌种,通常为500μl菌液+500μl60%甘油;将传至10次的重组汉逊酵母表达菌株接入5ml不含Zeocin抗性的YPD液体培养基中,于37℃、200rpm摇床培养至OD值达50后,以1∶1000的比例转接于5mlYPD液体培养基中,以此类推,在不含Zeocin抗性的YPD液体培养基中连续传5次。将稳定后的菌液涂布含10mg/mlG418的YPD平板,于32℃倒置培养2-4天,从各平板中挑取生长旺盛的单菌落进行拷贝数检测。实施例5:重组汉逊酵母细胞的外源多核苷酸拷贝数检测(1)酵母基因组DNA的提取及定量接种实施例4获得的生长旺盛的酵母菌株单菌落到5ml的YPD液体培养基中培养基,37℃培养16~24h;取2ml酵母培养液,室温4500g离心3min收集菌体;应用500μlSCED溶液(1mol/L山梨醇、10mmol/L柠檬酸钠、10mmol/LEDTA、10mmol/LDTT二硫苏糖醇)重悬菌体,并加入50mg玻璃珠充分震荡5min,加入50μl10mg/ml溶壁酶,37℃温浴1h;加入60μl的10%SDS,30μl的蛋白酶K,10μl的RNaseA酶,室温放置10分钟后在55℃的水浴中培养2h;加入350μl饱和酚及350μl氯仿,充分混合后于13000rpm,离心10分钟,收集分层后的上层液;加入等体积(约700μl)氯仿,于13000rpm,离心10分钟,收集分层后的上层液;往上层液中加入140μl的3mol/L乙酸钠溶液,轻轻混匀后再加入700μl异丙醇,混匀后室温放置5min,于13000rpm,离心10分钟。弃上清,加入1ml70%乙醇进行清洗,于13000rpm,离心10分钟,倒去上清,DNA沉淀置于室温放置30min后,加入100μlTE溶解。通过测定OD260进行基因组DNA的定量,并将线性化的片段稀释至100ng/μl,保存于-20℃冰箱中,备用。(2)Southern印迹方法进行外源多核苷酸拷贝数的定量a:探针制备本发明所应用的MOX探针申请号为201210021524.X的中国专利申请中描述的MOX探针,应用Roche公司的DIGDNALabelingandDetectionkit试剂盒(CatNo:11093657910)进行探针制备。具体步骤为:向EP小管中加入10μlMOX启动子区的PCR产物(200ng/μl),并用封口膜封上,沸水煮沸10分钟。立即放入预冷(-20℃)的无水乙醇小盒中(骤然冷却)。擦干管外壁乙醇,离心(约10s),依次加入5μl内毒素检查用水、2μl10xHexanucleotideMix、2μl10xdTPLabelingMixture、1μl7KlenowEnzyme(labelinggrade),混匀,封口膜封口。37℃水浴过夜,-20℃保存。b:Southern印迹应用北京美莱博医学科技有限公司的地高辛杂交检测试剂盒I(CatNo:DIGD-l10)及HyB高效杂交液(CatNo:Hyb-500)按照产品说明书进行Southern印迹检测。(3)不同重组汉逊酵母菌株的拷贝数比较Southern印迹检测结果(见图1)显示:筛选获得的HP/pRMHP2.1-6wt菌株、HP/pRMHP2.1-6sc菌株及HP/pRMHP2.1-6hp菌株的拷贝数接近,拷贝数均大于31但小于63。实施例6:HPV6L1蛋白的原核表达及兔多克隆抗体的制备(1)HPV6L1基因的PCR扩增及表达质粒克隆构建以pRMHP2.1-6hp为模板,以引物9与引物10扩增获得大小为1519bp的HPV6L1hp基因片段(不带终止密码子),同时在上游引入NdeI酶切位点,在下游引入SalI酶切位点;引物9:5’-ggaattccatATGTGGAGACCATCGGACTCTACTG-3’(SEQIDNO:13)引物10:5’-CCGGTCGACGCGCTTCGTCTTCGCTCTTTTCC-3’(SEQIDNO:14)通过NdeI+SalI双酶切将6hp基因片段克隆入NdeI+XhoI双酶切的pET43.1a载体(Novagen公司)中,将测序验证正确的重组质粒命名为pET43.1a-6hp。(2)HPV6L1蛋白在大肠杆菌中的诱导表达重组质粒pET43.1a-6hp转化大肠杆菌表达菌株BL21-CodonPlus(DE3)-RIPL,获得表达工程菌BL21-CodonPlus(DE3)-RIPL/pET43.1a-6hp。挑取单菌落接入含5mlLB液体培养基(含Amp50μg/ml)的试管中,37℃培养至OD600为0.6-0.9时加入IPTG至终浓度0.5mmol/L,诱导时间为5h,同时设置未诱导的对照管。收集菌液,SDS-PAGE检测重组蛋白的表达情况(图2A)。(3)HPV6L1蛋白的纯化样品准备:挑取表达工程菌BL21-CodonPlus(DE3)-RIPL/pET43.1a-6hp单菌落接入含5mlLB液体培养基(含Amp50μg/ml)的试管中,37℃培养过夜,次日按1%的接种量接入含400mlLB液体培养基(含Amp50μg/ml)的1L三角瓶中,至OD600约为0.8时,加入IPTG至终浓度0.5mmol/L,诱导5h后收集菌液并离心,将菌体沉淀按1∶10(g/ml)的比例重悬于PBS溶液,在冰浴下超声破菌(5s,5s,400W,60cycles),10000r/min离心10min离心收集沉淀。纯化过程(图2B):将超声沉淀按1∶10(g/ml)的比例加入溶液(100mM磷酸盐,500mMNaCl,20mM咪唑,8M脲,pH8.0)中充分搅拌溶解,10000r/min离心10min,收集上清0.45μm膜过滤。上样于ChelatingSepharoseFF层析介质,平衡液A为100mM磷酸盐,500mMNaCl,20mM咪唑,8M脲,pH8.0,洗脱液B为100mM磷酸盐,150mMNaCl,500mM咪唑,8M脲,pH8.0,洗脱步骤:先用20%B流洗杂蛋白,再以100%B洗脱目的蛋白。收集目的蛋白洗脱峰。透析去除咪唑。(4)兔多克隆抗体制备兔免疫及血清收集:用纯化的原核表达的HPV6L1蛋白免疫2只新西兰大耳兔,免疫方法采用背部多点注射法,免疫程序为第0、3、6、8周各免疫一次,免疫剂量均为250μg/次,抗原需经弗氏完全佐剂(FCA)或弗氏不完全佐剂(FIA)充分乳化后再进行注射,首次免疫应用FCA,后续免疫应用FIA,于第9周通过颈动脉采血并分离血清,每只兔可以分离血清40-60ml。抗体纯化(图2C):抗血清经0.45μm过滤后,上柱于层析介质rProteinASepharoseFF,进行亲和层析纯化。在pH中性条件下,抗体结合于层析介质上,用柠檬酸-柠檬酸钠缓冲液(pH4.0)洗脱,收集抗体洗脱峰,立即用1MTris.Cl,pH9.0中和到pH中性。实施例7:不同重组汉逊酵母菌株的表达研究(1)重组汉逊酵母菌株的诱导表达从平板中挑取重组汉逊酵母单菌落,接入含5ml的YPG液体培养基的小试管中,37℃培养24小时;将菌液转接于含30mlYPM诱导培养基的100ml三角瓶中,初始密度为OD600=1,于37℃摇床诱导72小时,每隔12小时于菌液中加入终浓度为0.5%的甲醇溶液(即0.15ml/瓶)。取25ml诱导后的菌液转移至50ml的离心管中,于10000rpm离心10min,弃上清;以50ml的细胞裂解重悬菌体沉淀,充分混匀后,在超声仪中按“功率60%、时间20min、开5s、关5s”的超声破碎程序进行菌体破碎,破碎过程中需保持冰浴;将超声破碎的菌液,于10000rpm离心10min,收集上清于-20℃冻存备用。(2)重组汉逊酵母菌株HPV6L1蛋白的表达检测应用Western印迹半定量方法进行HPV6L1蛋白表达水平的检测制胶及电泳:配制12%的聚丙烯酰胺凝胶,加入待检样品及标准品,上层胶以8V/cm的电压,分离胶以15V/cm的电压进行电泳,至溴酚蓝到达分离胶底部时,停止电泳,切下分离胶;半干法转膜:PVDF膜先用甲醇处理15秒,用去离子水漂洗3遍后再浸泡入正极液至少5min;电泳胶浸泡于负极液中,按顺序安装电转移装置,150mA的电流进行电转35-45min;转膜完成后,检查预染蛋白标志物条带是否被全部转移。封闭:用镊子将膜加入倒有20ml5%牛奶的自封袋中,挤出气泡,用塑料薄膜封口机封口。摇床慢摇封闭1小时或过夜(过夜应放入冰箱冷藏保存)。一抗孵育:将制备获得的兔抗HPV6L1蛋白多抗以1/200的比例加入盛有5%牛奶的自封袋中,混匀后把膜放入,挤出气泡,用塑料薄膜封口机封口。摇床慢摇孵育1.5小时。洗膜:将膜取出用TBST洗5次,每次约100ml,时间为5分钟。最后一次为10分钟。二抗孵育:将HRP标记的羊抗兔二抗以1/1000的比例加入盛有5%牛奶的自封袋中,混匀后把膜放入,挤出气泡,用塑料薄膜封口机封口。摇床慢摇孵育2小时。洗膜:将膜取出用TBST洗5次,每次约100ml,时间为5分钟。最后一次为10分钟。显色:将膜放入显色液中,摇床慢摇至条带出现,用流水冲洗终止显色,晾干后进行拍照。(3)结果通过比较不同HPV6L1核苷酸序列对于蛋白表达的影响,结果显示(图3):HPV天然基因6wt及按照酿酒酵母密码子偏爱性优化的6sc基因在汉逊酵母中的表达水平远低于按照汉逊酵母密码子偏爱性优化设计的基因6hp,因此,针对汉逊酵母宿主遗传密码偏爱性综合优化对于HPV6L1蛋白在汉逊酵母菌株中实现高水平表达是至关重要的。实施例8:HPV6L1重组汉逊酵母表达菌株的发酵工艺及纯化工艺研究发酵种子液:取1支冻存甘油菌(HP/pRMHP2.1-6hp)。融化后吸取80μl接入5mlYPD培养基中,于37℃、200rpm摇床培养18-24hr,A600nm约3-5,检定合格后各吸取1ml接入2瓶500mlYPD培养基中,于37℃、200rpm摇床培养18-24hr至A600nm约15-20,检定合格后作为发酵种子液待用。发酵控制:发酵起始培养基含有酵母粉300g,蛋白胨150g,甘油100g,基础盐(K2SO4273g,MgSO4100g,85%H3PO4400ml,KOH62g),10L纯化水充分溶解,加入30L发酵罐中,纯化水定容至14L,121℃,30min灭菌,冷却至30℃后加入60mlPTM1微量元素液(CuSO4·5H2O6.0g,K10.088g,MnSO4·H2O3.0g,Na2MoO4·2H2O0.2g,H3BO30.02g,CoCl2·6H2O0.5g,ZnCl220.0g,FeSO4·7H2O65.0g,Biotin0.2g,浓H2SO45.0ml,纯化水定容至1L,0.22μm滤膜过滤除菌),氨水调节pH5.6,接种2瓶500ml发酵种子液,此时发酵体积为15L。起始搅拌转速为200rpm、空气流量0.5Nm3/hr、罐压0.5bar,发酵中控制溶氧值为20-80%。起始生长期维持约22hr后,菌液A600nm达到20左右,溶氧开始快速上升,开始以100ml/hr流速流加补料培养基(50%甘油(W/V),12mlPTM1),此时进入流加生长期。培养约6-8hr后,菌液A600nm达到90左右,停止流加,氨水调节pH值至6.0。溶氧开始回升后开始流加甲醇(含12ml/LPTM1)进入诱导表达期,甲醇初始流加速率为50ml/hr,每小时取样,甲醇电极测定甲醇浓度,通过调节甲醇流加速率控制甲醇浓度<5g/L,并收集湿菌体超声破碎后用于Western印迹检测(样品5倍稀释),Western印迹检测结果如图4所示。下罐离心:诱导10hr后进行下罐,4℃条件下以5000rpm离心30min收集湿菌体。收获的湿菌体-20℃冻存。菌体破碎:取-20℃保存的HPV6表达湿菌体,按照10ml/g湿菌体的比例加入0.9%生理盐水清洗。菌体洗涤后,按20ml缓冲液/g湿菌体加入破碎缓冲液(含0.5mol/LNaCl,0.02%Tween-80,0.05mol/LMOPS)充分溶解,使用高压匀浆破碎,破碎压力1500bar,循环破碎6-8遍,镜检破碎率>90%。菌体破碎液在4℃条件下以9000rpm离心30min,收集上清液,再加入45%硫酸铵,沉淀30min后,4℃条件下9000rpm离心30min,收集上清液。层析纯化:经1μm过滤的菌体破碎上清液,上样于层析介质POROS50HS(AppliedBiosystems),目的蛋白吸附到层析介质上,以0.5M~1.5MNaCl梯度洗脱,目的蛋白与杂质获得初步分离,收集洗脱的HPV6L1蛋白(图5A)。将初纯的HPV6L1蛋白上样于层析介质Macro-Prep陶瓷羟基磷灰石(TypeII,40μm),目的蛋白结合于层析介质上,用20~200mM磷酸盐浓度梯度洗脱,目的蛋白与杂质分离,收集洗脱的HPV6L1蛋白(图5B)。HPV6L1VLP的解聚与重聚:纯化后的HPV6L1蛋白加入25mMDTT,0.05%的Polysorbate80,于pH8.2条件下,在室温解聚2小时。透析去除DTT,透析体系:1.0MNaCl,20mM磷酸盐,0.05%Polysorbate80,pH7.2。按1∶100比例透析,换3次,每次30min。再透析到重聚缓冲液中,透析体系:1MNaCl,5mMCa2+,60mM柠檬酸钠,pH6.2,0.05%Polysorbate80。于4℃,透析24小时后获得重聚的HPV6L1VLP。重聚前后HPV6L1蛋白的稳定性比较:1)热稳定性研究:在可控速率条件下,将温度由25℃逐渐提高至70℃,由于蛋白聚集导致的溶液中光散射发生变化,可通过350nm处的光吸收值进行检测。表1显示:未重聚的HPV6L1蛋白在50℃开始发生明显的聚集,而重聚后的HPV6L1蛋白在50℃时并未发生明显的聚集,加温至60℃才开始聚集。2)加速稳定性研究:在37℃条件下,比较放置一定时间后,未重聚的HPV6L1蛋白与重聚的HPV6L1蛋白的聚集程度,由表2可见,重聚后HPV6L1蛋白的加速稳定性明显好于未重聚的HPV6L1蛋白。综合热稳定性及加速稳定性研究结果,重聚提高了HPV6L1蛋白对于温度诱导聚集的内在稳定性。表1:重聚前后HPV6L1蛋白的热稳定性分析表2:重聚前后HPV6L1蛋白的加速稳定性分析实施例9:透射电镜观察纯化的HPV6L1重组蛋白用无菌水2倍稀释纯化后的HPV6L1蛋白样品,滴一小滴于腊盘上。取铜网使有支持膜的表面与样品液表面接触,静置1min取出,取出铜网,用滤纸条吸除多余的液滴,稍晾干。取2%醋酸铀溶液,滴一小滴于腊盘上。吸附有样品的铜网放置于染液表面(样品与染液接触),静置2min。取出铜网,用滤纸条吸除多余的液滴,白炽灯下晾干。应用JEOL-1400型号透射电镜观察VLP颗粒形态并拍照(结果图6所示)。实施例10:用汉逊酵母重组制备的HPV6L1VLP的免疫原性研究应用测定体液免疫效力ED50(半数有效剂量)的方法评价HPV6L1VLP的免疫原性(1)小鼠的免疫:70只6周龄Balb/c雌性小鼠(购自中国医学科学院实验动物研究所),清洁级饲养。共分为6个组,包括5个实验组及1个对照组,按所需免疫剂量将HPV6L1蛋白样品进行稀释(表3)。免疫程序为:第0、3、6周各免疫一次,最后一次免疫后14天杀鼠并分离血清。表3:小鼠的分组(2)ELISA法测定HPV6L1VLP免疫小鼠后的血清阳转率,具体步骤为:用包被缓冲液将大肠杆菌重组HPV6L1蛋白稀释至0.5μg/ml,每孔加0.1ml,4℃过夜。次日洗涤缓冲液洗涤3次,甩尽残余液体。以抗体稀释液封闭30分钟,洗涤缓冲液洗涤3次,甩干后作检测,或晾干后4℃防湿保存。用样品稀释液将各小鼠血清样品以1∶10000进行稀释,取0.1ml于上述已包被之反应孔中,置37℃孵育1小时,洗涤5次。(同时做空白、阴性孔对照)。于反应孔中加入抗体稀释液1∶5000新鲜稀释的HRP标记的羊抗鼠IgG第二抗体0.1ml,37℃孵育30分钟,洗涤5次,最后一遍用双蒸水洗涤。于各反应孔中加入临时配制的TMB底物溶液0.1ml,37℃显色10分钟。于各反应孔中加入50μl2M硫酸0.05ml以终止反应。在酶标仪上,于450nm处(630nm为参考波长),以空白对照孔调零后测各孔OD值。Cutoff值计算及阳性结果判定:Cutoff值=阴性对照值×2.1;样品OD值>Cutoff值则判为阳性。(3)体液免疫效力ED50的计算根据表4计算结果,HPV6L1VLP的体液免疫效力ED50值为0.315μg,显示了HPV6L1VLP具备良好的免疫原性。表4:小鼠的体液免疫效力检测情况其中:高于50%阳转稀释度为2,低于50%阳转稀释度为4。通过计算获得:距离比为0.667,log10高于50%阳转的稀释度为0.301,ED50的对数为0.5017,50%阳转稀释度为3.17。所以,ED50值为0.315μg。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1