具有三嗪‑哌嗪主链的α‑螺旋类似物及其制备方法与流程
本发明涉及作为α-螺旋模拟物的具有三嗪-哌嗪支架的化合物。所述化合物模拟蛋白质二级结构的α-螺旋结构,并且充当由α-螺旋介导的对多种疾病具有特异性的蛋白质相互作用的抑制剂。因此,所述化合物可用作生物探针并且具有用于疾病的治疗剂的高潜力。
背景技术:
细胞通过进行多种生物学功能(例如基因调节、细胞生长、细胞周期、代谢和通过多样且复杂的蛋白质-蛋白质相互作用(PPI)的信号转导)来维持生命机能。此外,已知许多疾病与不正常的蛋白质-蛋白质相互作用或蛋白质错误折叠相关。理解细胞中的蛋白质-蛋白质相互作用及其功能是理解生命现象的基石并且是开发新药和治疗疾病的重要基础。
在许多蛋白质相互作用中,已知蛋白质通过识别2至3圈的短α-螺旋与靶蛋白结合。α-螺旋结构具有约3.14个氨基酸残基的一个旋转(圈)和对于每3至4个残基空间布置的侧链。由侧链排列的空间被称为i、i+3/i+4、i+7等,位于i、i+3/i+4和i+7中的氨基酸残基充当与靶蛋白结合的识别基序。因此,可模拟α-螺旋肽的结构的低分子物质预期起到蛋白质相互作用抑制剂的作用,并且已积极地进行关于开发α-螺旋模拟物的研究。
关于这一点,已经报道了具有三联苯支架的α-螺旋样化合物(J.Am.Chem.Soc.2001,123,5382-3;Nat.Chem.2013,5(3):161-173;Curr.Opin.Chem.Biol.2010,14(3):341-346)。如上所述,α-螺旋本质上具有用作蛋白质结合的识别基序的三个氢基酸残基(在位置i、i+3/i+4、i+7处的氢基酸残基)。通过使分别连接至构成三联苯支架的三个苯环的取代基位于对应于三个氨基酸残基的三维空间位置,将具有三联苯支架的α-螺旋模拟化合物设计成用于模拟α-螺旋的功能。
然而,基于三联苯结构的α-螺旋模拟物在开发为治疗剂方面具有许多困难,因为其不仅难以合成,而且由于高疏水性在水溶液中具有低溶解度。
技术实现要素:
技术问题
因此,必须开发具有这样的结构的低分子α-螺旋模拟物:其在水溶液中具有良好的溶解度、细胞渗透性并且容易合成。
技术方案
本发明人已经开发了具有三嗪-哌嗪作为核心结构的新α-螺旋模拟化合物以克服具有三联苯支架的α-螺旋模拟物的问题。还证实了,本发明的α-螺旋模拟化合物成功地模拟了α-螺旋所必需的三个氨基酸残基(i、i+3或i+4、i+7)的三维取向。此外,本发明人已经开发了用于简单合成具有三嗪-哌嗪支架的α-螺旋模拟物的固相合成法。
因此,本发明的一个目的在于提供具有三嗪-哌嗪作为核心结构的α-螺旋模拟化合物及其盐。
本发明的另一个目的在于提供用于制备α-螺旋模拟化合物的方法。
本发明的另一个目的在于提供用于抑制蛋白质相互作用的组合物,其包含α-螺旋模拟化合物或其盐。
本发明的另一个目的在于提供用于抑制蛋白质相互作用的方法,其包括处理α-螺旋模拟化合物或其盐。
本发明的另一个目的在于提供用检测蛋白质相互作用的组合物,其包含α-螺旋模拟化合物或其盐。
本发明的另一个目的在于提供用于治疗与α-螺旋介导的蛋白质相互作用相关的疾病的药物组合物,其包含α-螺旋模拟化合物或其盐。
本发明的另一个目的在于提供用于治疗与α-螺旋介导的蛋白质相互作用相关的疾病的方法,其包括向个体施用有效量的α-螺旋模拟化合物或其盐。
有益效果
本发明提供了具有三嗪-哌嗪支架的α-螺旋模拟化合物。所述化合物可通过高效地模拟蛋白质二级结构的α-螺旋结构来用作对多种疾病具有特异性的α-螺旋介导的蛋白质相互作用的抑制剂。
附图说明
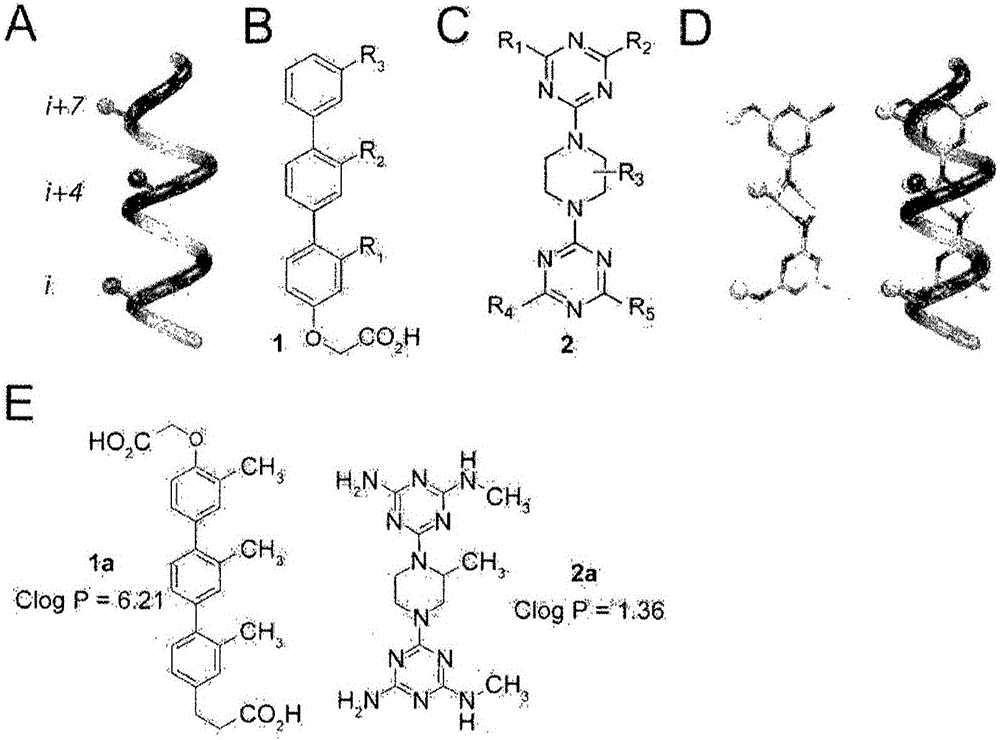
在图1中,A表示α-螺旋中的侧链i、i+4和i+7的位置,B表示具有三联苯支架的常规α-螺旋模拟物的结构,C表示本发明的具有三嗪-哌嗪-三嗪支架的α-螺旋模拟物的结构,D示出了当本发明的α-螺旋模拟物制备为能量最小化结构(2a)(即,R2、R3、R5=CH3;R1、R4=H)时与α-螺旋叠加,并且图1E为当本发明的α-螺旋模拟物和具有三联苯支架的常规α-螺旋模拟物制备为能量最小化结构(1a、1b)时Clog P值的对比结果。
图2表示用于制备具有三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的固相合成法的概况。
图3表示使用类肽作为编码标签来构建α-螺旋模拟化学库的过程。
图4表示从α-螺旋模拟化学库中选择与靶蛋白结合的α-螺旋模拟化合物的过程。
图5表示用荧光材料标记化合物9a、9b和9c的过程。
在图6中,A表示通过与MCL-1结合来选择的化合物9a(化学式4)、9b(化学式5)和9c(化学式6)以及阴性对照9-nc的结构,B表示荧光偏振测定的结果,该结果表明化合物9a、9b和9c与MCL-1结合,并且C表示比较荧光偏振测定的结果,该结果表明化合物9a、9b和9c以及荧光标记的BH-3肽与MCL-1结合。
在图7中,A示出了MCL-1和BH-3肽复合物的晶体结构,表明MCL-1的主要结合部位为BH3的三个疏水残基(Leu213、Val216和Val220)的侧链。B为计算机对接测定的结果,该结果表明化合物9c与MCL-1的BH3结合部位结合。
在图8中,A表示MCL-1免疫沉淀和蛋白质印迹的结果,该结果表明Jurkat T细胞中的MCL-1/BAK相互作用在经不同浓度的化合物9c(10μM、50μM、100μM)或DMSO处理2.5小时后受到抑制。B示出了在用1μM的9c-FL或5(6)-羧基荧光素(Flu)处理A549细胞4小时之后的共聚焦显微术结果。C示出了经不同浓度的化合物9c和9-nc(阴性对照)处理的Jurkat T细胞(左图)和WS-1正常成纤维细胞(右图)的细胞生存力(%)。
图9示出了MCL-1免疫沉淀和蛋白质印迹的结果,该结果表明在用化合物9c(25μM、50μM)、化合物9-nc(100μM)或DMSO处理Jurkat T细胞2.5小时之后MCL-1/BAK相互作用受到抑制。
在图10中,A表示用不同浓度的化合物9c和9-nc(阴性对照)处理24小时的U266细胞(左图)的细胞生存力(%)。B表示用不同浓度的化合物9c和9-nc(阴性对照)处理24小时的正常细胞的细胞生存力(%)。C示出了在指定浓度的DMSO下用化合物9c处理U266细胞和WS-1细胞16小时之后的胱天蛋白酶活性分析的结果。
图11表示用多种亲水基团取代对应于化合物9c的R1位置的Et的过程。
图12表示用多种亲水基团取代对应于化合物9c的R4位置的氢的过程。
在图13中,A表示化合物Q1(化学式7)和化合物Q2(化学式8)的结构。B表示荧光偏振测定的结果,该结果表明每种化合物Q1、Q2、9a、9b、9c和作为阴性对照的氟接头与野生型α-突触核蛋白结合。C示出了在有或无化合物Q1和Q2的情况下监测α-突触核蛋白的去折叠的CD(圆二色性(circular dichroism))热变性的结果。
在图14中,A表示荧光偏振测定的结果,该结果表明每种化合物Q1、Q2、9a、9b、9c和氟接头与突变体α-突触核蛋白A53T结合。B表示硫磺素T聚集测定的结果,该结果表明每种化合物Q1、Q2和阴性对照9c影响突变体α-突触核蛋白A53T的聚集。
图15表示制备具有苯基-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的固相合成法的概况。(a)示出了4-氟-3-硝基苯甲酸、HATU、DIEA和DMF在室温下反应24小时;(b)示出了N-硝基苯磺酰基保护的哌嗪衍生物(“3”)、DIEA和DMF在95℃下反应12小时;(c)示出了2-巯基乙醇、DBU和DMF在室温下反应3小时;(d)示出了2-乙基氨基-4,6-二氯-[1,3,5]三嗪(“5”)、DIEA和THF在60℃下反应3小时;(e)示出了R3NH2、DIEA和NMP在80℃下反应过夜;(f)示出了SnCl2·2H2O和DMF在室温下反应24小时;(g)示出了在条件A(芳香醛)的情况下R1CHO、CH(OMe)3/DMF/MeOH 9∶1∶2(1%HOAc)在50℃下反应18小时,然后NaCNBH3和THF在50℃下反应6小时;以及在条件B(脂肪醛)的情况下R1CHO、苯并三唑、CH(OMe)3/DMF/MeOH 9∶1∶2(1%HOAc)在室温下反应18小时,然后NaCNBH3和THF在室温下反应6小时;以及(h)示出了95%TFA在CH2Cl2中在室温下反应3小时。
图16示出了竞争性荧光偏振测定的结果,该结果表明36种具有苯基-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物抑制Mcl-1与BH3肽之间的相互作用。蓝柱示出了10μM下的抑制作用而红柱示出了50μM下的抑制作用。
在图17中,A示出了指定为PPT-31的化学式10化合物的结构,B示出了与BH3螺旋肽和能量最小化结构叠加的分子模拟的结果。C示出了PPT-31的抑制曲线,其中荧光标记的BH3肽抑制Mcl-1(黑色)与Bcl-XL(灰色)的结合。误差条是指三次独立测试结果的标准偏差。
具体实施方式
本发明涉及具有三嗪-哌嗪作为核心结构的α-螺旋模拟化合物及其盐,以及其用途。
根据本发明,具有三嗪-哌嗪作为核心结构的α-螺旋模拟化合物可如以下化学式1所示:
[化学式1]
在化学式1中,
X为
R1、R2、R4和R5独立地为-NR8R9或-OR10,
R8和R9独立地为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环基、-N-(C3-C10)芳基、-N-(C3-C10)杂芳基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)m-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)m-(C3-C10)杂环、-(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,其中m为0至3的整数,
R8和R9可彼此连接以与C、N或O一起形成环,
R10为氢、可被独立地选自C1-8烷基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、羧基、羟基、氨基、氧代、苯基和苯氧基的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)n-(C3-C10)环烷基、-(CH2)n-(C3-C10)杂环、-(CH2)n-(C3-C10)芳基或-(CH2)n-(C3-C10)杂芳基,其中n为0至3的整数,
R3为(C1-C6)烷基或-(CH2)p-(C3-C10)芳基,其中p为0至3的整数,
R6为-NR11,其中R11为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个的取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)q-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)q-(C3-C10)杂环、-(CH2)q-(C3-C10)芳基或-(CH2)q-(C3-C10)杂芳基,其中q为0至3的整数,
R11可彼此连接以与C或O一起形成环,并且
R7为-NH2。
本发明的化合物包括立体异构体,例如具有所有基本分子结构的非对映体和对映体。特别地,手性中心可具有(R)-或(S)-构型,二价环状(部分地)饱和基团上的取代基可具有顺式或反式构型。包含碳-碳双键的化合物可在双键处具有E或Z-记号。化学式1中所示的化合物的立体异构体旨在包括在本发明的范围内。
具体地,本发明的化合物包括具有三嗪-哌嗪-三嗪作为核心结构的化合物和具有苯基-哌嗪-三嗪作为核心结构的化合物。
在下文中,将更详细地描述本发明提供的化合物。
在一个实施方案中,本发明涉及具有三嗪-哌嗪-三嗪的核心结构的α-螺旋模拟化合物及其盐。
具有三嗪-哌嗪-三嗪的核心结构的化合物可如以下化学式2所示:
[化学式2]
在化学式2中,
R1、R2、R4和R5独立地为-NR8R9或-OR10,
R8和R9独立地为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环、-N-(C3-C10)芳基、-N-(C3-C10)杂芳基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)m-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)m-(C3-C10)杂环、-(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,其中m为0至3的整数,
R8和R9可彼此连接以与C、N或O一起形成环,
R10为氢、可被独立地选自C1-8烷基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、羧基、羟基、氨基、氧代、苯基和苯氧基的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)n-(C3-C10)环烷基、-(CH2)n-(C3-C10)杂环、-(CH2)n-(C3-C10)芳基或-(CH2)n-(C3-C10)杂芳基,其中n为0至3的整数,
R3为(C1-C6)烷基或-(CH2)p-(C3-C10)芳基,并且
p为0至3的整数。
在一些具体的实例中,在化学式2中,R1、R2、R4和R5独立地为-NR8R9,在R1和R4的情况下,R8和R9独立地为氢、可被C1-4烷氧基、羟基或苯氧基取代的(C1-C6)烷基、-(CH2)rNH2、-(CH2)rN(H)C(=NH)NH2或-(CH2)rC(=O)OH或-(CH2)rOH,并且r为1至6的整数;或者在R2和R5的情况下,R8和R9独立地为氢、可被C1-4烷氧基、羟基或苯氧基取代的(C1-C6)烷基、-(CH2)m-(C3-C10)环烷基、(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,并且m为0至3的整数,并且R3为(C1-C6)烷基或苄基。
在另一个实例中,在化学式2中,R1、R2、R4和R5独立地为-NR8R9,在R1和R4的情况下,R8和R9独立地为氢或(C1-C6)烷基,或者在R2和R5的情况下,R8和R9独立地为C1-C6烷基、-(CH2)m-苯基、-(CH2)m-苯氧基苯基、-(CH2)m-苯并二氧杂环戊烯、-(CH2)m-二烷氧基苯、-(CH2)m-二甲氧基苯基、-(CH2)m-羟基苯基或-(CH2)m-(C3-C6)环烷基,并且R3为(C1-C6)烷基或苄基。
在另一个实例中,在化学式2中,R1、R2、R4和R5独立地为-NR8R9,在R1和R4的情况下,R8和R9独立地为氢或(C1-C6)烷基,或者在R2和R5的情况下,R8和R9独立地为C1-C6烷基、-(CH2)m-苯基、-(CH2)m-苯氧基苯基、-(CH2)m-苯并二氧杂环戊烯、-(CH2)m-二烷氧基苯、-(CH2)m-二甲氧基苯基、-(CH2)m-羟基苯基或-(CH2)m-(C3-C6)环烷基,并且R3为(C1-C6)烷基或苄基。
作为另一个实施方案,化学式2中的R1、R2、R4和R5独立地为选自下列基团中的至少一种:
作为另一个实施方案,化学式2中的R3独立地为选自下列基团中的至少一种:
作为另一个实施方案,化学式2的化合物和化学式4至化学式8的化合物为选自下列组中的至少一种:
[化学式4]
[化学式5]
[化学式6]
[化学式7]
[化学式8]
作为另一个实施方案,本发明的化合物可以是化学式9的化合物:
[化学式9]
在化学式9中,
R1和R4独立地为-NR8R9或-OR10,
R8和R9独立地为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环基、-N-(C3-C10)芳基、-N-(C3-C10)杂芳基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)m-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)m-(C3-C10)杂环、-(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,并且m为0至3的整数,
R8和R9可彼此连接以与C、N或O一起形成环,并且
R10为氢、可被独立地选自C1-8烷基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、羧基、羟基、氨基、氧代、苯基和苯氧基的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)n-(C3-C10)环烷基、-(CH2)n-(C3-C10)杂环、-(CH2)n-(C3-C10)芳基或-(CH2)n-(C3-C10)杂芳基,其中n为0至3的整数。
或者,在化学式9中,R1和R4独立地为-NR8R9,
R8和R9独立地为氢、(C1-C6)烷基、-(CH2)rNH2、-(CH2)rN(H)C(=NH)NH2、-(CH2)rC(=O)OH或-(CH2)rOH,并且
r为1至6的整数。
作为本发明的另一个实施方案,提供了包含三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物及其盐。包含苯基-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物如化学式3所示:
[化学式3]
在化学式3中,
R1和R2独立地为-NR8R9或-OR10,
R8和R9独立地为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环基、-N-(C3-C10)芳基、-N-(C3-C10)杂芳基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)m-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)m-(C3-C10)杂环、-(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,其中m为0至3的整数,
R8和R9可彼此连接以与C、N或O一起形成环,
R10为氢、可被独立地选自C1-8烷基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、羧基、羟基、氨基、氧代、苯基和苯氧基的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)n-(C3-C10)环烷基、-(CH2)n-(C3-C10)杂环、-(CH2)n-(C3-C10)芳基或-(CH2)n-(C3-C10)杂芳基,其中n为0至3的整数,
R3为(C1-C6)烷基或-(CH2)p-(C3-C10)芳基,p为0至3的整数,
R6为-NR11,其中R11为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)q-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)q-(C3-C10)杂环、-(CH2)q-(C3-C10)芳基或-(CH2)q-(C3-C10)杂芳基,并且q为0至3的整数,
R11可彼此连接以与C或O一起形成环,并且
R7为-NH2。
在一个具体的实例中,在化学式3中,
R1和R2独立地为-NR8R9,其中R8为氢,R9为氢、C1-C6烷基或-(CH2)m-(C3-C10)芳基,并且m为0至3的整数,
R3为(C1-C6)烷基或苄基,
R6为-NR11,其中R11为(C1-C6)烷基或-(CH2)q-(C3-C10)芳基并且q为0至3的整数,并且
R7为-NH2。
在另一个实施方案中,R1和R2独立地选自如以下化学结构所示化合物:
在另一个实施方案中,化学式3的R3独立地选自如以下化学结构所示基团:
在另一个实施方案中,化学式3的R6为选自下列基团的至少一种:
在另一个实施方案中,化学式3的R1、R2、R3、R6和R7独立地为选自下列基团的至少一种:
[表1]
在另一个实施方案中,化学式3的化合物具体地为化学式10的化合物:
[化学式10]
在本发明的化合物中,R2、R3、R5和R6位于对应于三个氨基酸(在i、i+3/i+4、i+7处的氨基酸残基)的三维空间中,所述氨基酸充当与蛋白质结合的天然α-螺旋的识别基序,从而允许α-螺旋模拟化合物与靶蛋白结合。
此外,R1和R4可优选地包含亲水基团,但不限于此。当R1和R4可包含亲水基团时,α-螺旋模拟化合物为两亲性的。大部分天然α-螺旋肽在一侧上包含疏水残基并且当与其他蛋白质结合时作为识别基序发挥重要作用,而α-螺旋肽在相对侧上包含亲水氨基酸残基并且当α-螺旋肽与蛋白质连接时,这些亲水残基朝外并位于暴露区域中。因此,α-螺旋肽处于能量上有利的状态。当α-螺旋肽与靶蛋白结合时,α-螺旋肽的亲水残基与靶蛋白表面上的氨基酸具有亲水相互作用。除了疏水相互作用之外,亲水相互作用可使α-螺旋肽与靶蛋白更强地结合。因此,作为一个优选的实施方案,本发明提供具有亲水取代基和疏水取代基的α-螺旋模拟化合物,并且模拟天然α-螺旋的两亲性质。迄今为止开发的α-螺旋模拟化合物几乎没有具有两亲性质的α-螺旋模拟化合物。
因此,作为一个优选的实施方案,本发明的α-螺旋模拟化合物包含三嗪-哌嗪作为核心结构,并且三个取代基(R2、R3、R5或R2、R3、R6)主要通过疏水相互作用在与靶蛋白结合中发挥重要作用,并且位于三个取代基对侧的两个取代基(R1、R4)包含亲水基团并模拟天然α-螺旋的两亲性质,从而被设计成蛋白质-蛋白质相互作用的强抑制剂。根据本发明的α-螺旋模拟化合物的性质概略地在图1中示出。
如图1中所示,当在最低能量状态下合成本发明的具有三嗪-哌嗪-三嗪支架的化合物和具有三联苯支架的常规化合物时,本发明化合物和常规化合物的Clog P(计算的log P辛醇/水)分别为1.366和6.21。因此,本发明的化合物表现出更加改善的亲水性,从而实现与药物相似的性质。
在本文中取代基的定义中,术语“烷基”是指脂肪族烃基。烷基可以是“饱和烷基”(其不包含烯基或炔基部分)或“不饱和烷基”(其包含至少一个烯基或炔基部分)。“烯基”意指包含至少一个碳-碳双键的基团,并且“炔基”意指包含至少一个碳-碳三键的基团。当单独使用或与烷氧基组合使用时,烷基可以是支链或直链。
除非另有限定,否则烷基可包含1至20个碳原子。烷基可以是具有1至10个碳原子的中等大小的烷基。烷基可以是具有1至6个碳原子的低级烷基。典型的烷基包括但不限于:甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基、乙烯基、丙烯基、丁烯基等。例如,C1-C4烷基在烷基链中包含1至4个碳原子并且选自:甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基和叔丁基。
除非另有限定,否则术语“烷氧基”意指具有1至10个碳原子的烷氧基。例如,烷氧基可以是甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基、异丁氧基、仲丁氧基等。
除非另有限定,否则术语“环烷基”意指饱和的脂肪族3元至10元环。典型的环烷基包括但不限于环丙基、环丁基、环戊基、环己基等。
术语“芳基”包括至少一个具有共价π电子系统的环并且包括例如单环或稠环多环基团(即,共享相邻的碳原子对的环)。即,在本文中,除非另有限定,否则芳基可意指4元至10元,优选6元至10元芳香族单环或多环的环,例如苯基、萘基等。
术语“杂芳基”意指包含1至3个选自N、O和S(其可与苯并或C3至C8环烷基稠合)的杂原子的芳香族3元至10元,优选4元至8元环,更优选5元至6元环。单环杂芳基的实例包括噻唑、唑、噻吩、呋喃、吡咯、咪唑、异唑、异噻唑、吡唑、三唑、三嗪、噻二唑、四唑、二唑、吡啶、哒嗪、嘧啶、吡嗪和类似的基团。环状杂芳基的实例包括但不限于:吲哚、二氢吲哚、苯并噻吩、苯并呋喃、苯并二氧杂环戊烯、苯并咪唑、苯并唑、苯并异唑、苯并噻唑、苯并噻二唑、苯并三唑、喹啉、异喹啉、嘌呤、嘌呤吡啶和类似的基团。
除非另有限定,否则术语“杂环”意指包含1至3个选自N、O和S(能够与苯并或C3至C8环烷基稠合并且为饱和的或者1或2个双键)的杂原子的3元至10元环,优选4元至8元环,更优选5元至6元环。杂环的实例包括吡咯啉、吡咯烷、咪唑啉、咪唑烷、吡唑啉、吡唑烷、吡喃、哌啶、吗啉、硫代吗啉、哌嗪和氢呋喃,但不限于此。
术语“芳基烷基”意指可进一步被芳基取代的C1-C4亚烷基,例如-CH2-、-CH2CH2-等。芳基烷基的实例为苄基、苯乙烯等。
术语“环烯基”是指包含4至10个环碳和一个或更多个双键的非芳族环基团。一些实例包含4至6个碳,而另一些实例包含4或5个碳,而一些实施方案包含4个碳。环烯基的实例包括环丁烯基、环戊烯基、环己烯基等。
术语“环烷基”是指包含3至10个碳的饱和环基团,一些实施方案包含3至6个碳,一些实施方案包含3至5个碳,一些实施方案包含5至7个碳,而一些实施方案包含3或4个碳。实例包括环丙基、环丁基、环戊基、环己基、环庚基等。
术语“卤素”或“卤代”意指氟、氯、溴或碘。
术语“卤代烷基”是指如本文中所限定的C1-6烷基,其中烷基被一个卤素取代和完全被卤素取代。完全取代的C1-6卤代烷基如化学式CnL2n+1所示,其中L为卤素,并且N为1、2、3、4、5或6。具有一个或更多个卤素原子的卤代烷基可以相同或不同并且选自F、Cl、Br和I。卤代烷基的实例包括但不限于氟甲基、二氟甲基、三氟甲基、氯二氟甲基、2,2,2-三氟乙基、五氯乙基等。
术语“氨基”表示-NH2基团。
术语“苄基”是指-CH2C6H5。
术语“羧基”是指-CO2H基团并且可被称为羧酸。
术语“羟基”是指-OH基团。
术语“硝基”是指-NO2基团。
在本文中,术语“氧代”取代基是指=O,并且当碳被氧代基团取代时,由碳和氧代产生羰基。
术语“苯氧基”是指C6H5O-基团。
术语“苯基”是指C6H5-基团。
术语“磺酰胺”是指-SO2NH2基团。
除非另有限定,否则本文中使用的其他术语和缩写应理解为本发明所属领域的技术人员通常理解的含义。
根据本发明的化合物还可形成可药用盐。
这样的术语“可药用盐”可包括酸形成的包含可药用阴离子的无毒酸加成盐,例如由以下酸所形成的酸加成盐:无机酸,例如硫酸、盐酸、硝酸、磷酸、氢溴酸、氢碘酸等;有机羧酸,例如酒石酸、甲酸、柠檬酸、乙酸、三氯乙酸、三氟乙酸、葡糖酸、苯甲酸、乳酸、富马酸、马来酸、水杨酸等;磺酸,例如甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸、萘磺酸等。此外,所述盐包括可药用碱加成盐,例如由锂、钠、钾、钙、镁等形成的碱金属盐或碱土金属盐;氨基酸盐,例如赖氨酸、精氨酸和胍;有机盐,例如二环己基胺、N-甲基-D-葡糖胺、三(羟甲基)甲胺、二乙醇胺、胆碱、三乙胺等。根据本发明的化合物可通过一般方法转化成其盐,并且本领域技术人员基于化学式的结构可以容易地进行盐的制备而无需进一步解释。
在另一个实施方案中,本发明涉及用于制备具有三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的方法。
例如,本发明的方法可包括以下步骤:
通过将化学式(a)的化合物与经胺(NH2)修饰的聚合物珠连接来制备化学式(b)的化合物,
通过将化学式(b)的化合物与被2-硝基苯磺酰基保护的化学式(c)的化合物偶联来制备化学式(d)的化合物,
在从化学式(d)的化合物中除去2-硝基苯磺酰基保护基之后,通过引入化学式(e)的化合物来制备化学式(f)的化合物,
用胺基团(R2”R2”NH)取代如化学式(f)所示化合物的氯位置,以及
除去经胺(NH2)修饰的聚合物珠。
[化学式a]
[化学式b]
[化学式c]
[化学式d]
[化学式e]
[化学式f]
如制备方法中所提供的,本发明的α-螺旋模拟化合物可由非常简单的固相合成来制备。可通过经胺修饰的聚合物珠中的若干个顺序偶联反应步骤以92%的平均收率获得目的化合物。通过参照图2对制备方法进行更详细地描述。作为第一步,可将单取代的二氯三嗪(“3”)加载到Rink-Amide MBHA树脂上以与树脂结合,从而产生与树脂结合的三嗪衍生物(“4”)。作为第二步,可通过将用2-硝基苯磺酰基(Ns)保护的哌嗪衍生物(“5”)与附着到树脂上的三嗪衍生物(“4”)偶联来制备化合物“6”。作为第三步,在从偶联的化合物(“6”)中除去N-Ns保护基之后,可通过向去保护的化合物中引入2-乙基氨基-4,6-二氯-[1,3,5]三嗪(“7”)来制备化合物(“8”)。作为第四步,可将多种胺(R2R2’H)引入到所制备化合物(“8”)的氯位点。作为第五步,可通过添加TFA(三氟乙酸)进行切割反应来制备用三个取代基官能化的化合物(“9”)。
在另一个方面,本发明涉及用于制备具有苯基-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的方法。
通过将4-氟-3-硝基苯甲酸与经胺(NH2)修饰的聚合物珠连接来制备化学式(b’)的化合物,
通过将化学式(b’)的化合物与由2-硝基苯磺酰基保护的化学式(c’)的化合物偶联来制备化学式(d’)的化合物,
在从化学式(d’)的化合物中除去2-硝基苯磺酰基保护基之后,通过引入化学式(e’)的化合物来制备化学式f’的化合物,
通过用胺基团(R3NH)取代化学式(f’)的化合物的氯并使硝基还原来制备化学式(g’)的化合物,
通过将R1官能团引入到化学式(g’)的化合物中来制备化学式(h’)的化合物,以及
除去经胺(NH2)修饰的聚合物珠。
[化学式b’]
[化学式c’]
[化学式d’]
[化学式e’]
[化学式f’]
[化学式g’]
[化学式h’]
如制备方法中所提供的,本发明的α-螺旋模拟化合物可通过非常简单的固相合成来制备。可通过经胺(NH2)修饰的聚合物珠中的若干个顺序偶联反应步骤以90%的平均收率获得目的化合物。
在另一个方面,本发明涉及用于抑制蛋白质-蛋白质相互作用的组合物,其包含α-螺旋模拟化合物或其盐。
在另一个方面,本发明涉及用于抑制蛋白质-蛋白质相互作用的方法,其包括用靶蛋白处理α-螺旋模拟化合物或其盐的步骤。
在另一个方面,本发明涉及用于检测蛋白质-蛋白质相互作用的组合物,其包含α-螺旋模拟化合物或其盐。
在另一个方面,本发明涉及用于检测蛋白质-蛋白质相互作用的方法,其包括用靶蛋白处理α-螺旋模拟化合物或其盐的步骤。
本发明提供的α-螺旋模拟化合物可模拟天然存在的α-螺旋肽的结构并且有效地模拟α-螺旋肽的功能,并因此可用作蛋白质-蛋白质相互作用的强抑制剂。其还可以利用与靶蛋白结合的特性用作检测蛋白质-蛋白质相互作用的探针。
在本发明中,提供了包含本发明的α-螺旋模拟化合物的化学库。所述库包含具有三嗪-哌嗪作为核心结构的两亲性α-螺旋模拟化合物,并且可基于靶蛋白的特性筛选与靶蛋白特异性地结合的化合物。靶蛋白可以是但不限于抗体、酶、受体或细胞系。
在本发明中,蛋白质与蛋白质之间的相互作用包括蛋白质与蛋白质之间的结合或蛋白质与蛋白质之间的聚集。这种相互作用可以例如在两种膜结合蛋白之间、在膜结合蛋白与胞质蛋白(例如,受体和配体)之间、在胞质蛋白之间等。优选地,本发明可通过α-螺旋结构应用于蛋白质-蛋白质相互作用。
在本发明的一个实施方案中,提供了抑制和检测MCL-1(髓样细胞白血病-1(myeloid cell leukemia-1))蛋白与BCL-2(B细胞淋巴瘤2(B-cell lymphoma 2))蛋白之间的相互作用(即,蛋白质-蛋白质结合)以及α-突触核蛋白之间的相互作用(即,蛋白质-蛋白质聚集)的α-螺旋模拟化合物的筛选,但这仅是一个代表性实例,并且本发明的范围不限于此。
在另一方面,本发明涉及用于治疗由α-螺旋介导之疾病的药物组合物,其包含α-螺旋模拟化合物或其盐。
在另一个方面,本发明涉及用于治疗与α-螺旋介导的蛋白质之间相互作用相关的疾病的方法,其包括向所述个体施用有效量的所述α-螺旋模拟化合物或其盐。
例如,本发明涉及用于预防或治疗癌症的药物组合物,其包含选自化学式4至化学式6以及化学式9和化学式10的化合物的至少一种化合物或其可药用盐。在另一个实例中,本发明涉及用于治疗癌症的方法,其包括向所述个体施用有效量的选自化学式4至化学式6、化学式9和化学式10的化合物的所述α-螺旋模拟化合物或其盐:
[化学式4]
[化学式5]
[化学式6]
[化学式9]
[化学式10]
在化学式中,
R1和R4独立地为-NR8R9或-OR10,
R8和R9独立地为氢、可被独立地选自C1-8烷基、C2-6烯基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、C3-C10杂芳基、C3-C10杂环基、-N-(C3-C10)芳基、-N-(C3-C10)杂芳基、羧基、胍基、羟基、硝基、氨基、苯基、苯氧基、氧代和磺酰胺的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)m-(C3-C10)环烷基、-(CH2)m-(C4-C10)环烯基、-(CH2)m-(C3-C10)杂环、-(CH2)m-(C3-C10)芳基或-(CH2)m-(C3-C10)杂芳基,并且m为0至3的整数,
R8和R9彼此连接以与C、N或O一起形成环,
R10为氢、可被独立地选自C1-8烷基、C1-4烷氧基、C3-6环烷基、卤素、C1-4卤代烷基、羧基、羟基、氨基、氧代、苯基和苯氧基的至少1至5个取代基取代的(C1-C6)烷基、(C2-C6)烯基、(C2-C6)炔基、-(CH2)n-(C3-C10)环烷基、-(CH2)n-(C3-C10)杂环、-(CH2)n-(C3-C10)芳基或-(CH2)n-(C3-C10)杂芳基,并且n为0至3的整数。
MCL-1(髓样细胞白血病-1)是具有抗凋亡活性的BCL-2(B细胞淋巴瘤2)家族蛋白之一。MCL-1抑制由BH3引起的细胞凋亡并且通过与BH3(BCL-2同源结构域3)家族蛋白(例如BAK)相互作用来帮助细胞存活。因此,抑制癌细胞中MCL-1蛋白与BH3蛋白之间的相互作用可防止癌细胞存活,从而是有前景的抗癌治疗策略。
在本发明中,为了证明三嗪-哌嗪-三嗪化合物可用作抑制蛋白质间相互作用的α-螺旋模拟物,抑制BH3蛋白与MCL-1蛋白之间的相互作用的化合物可选自本发明的库。为了选择抑制MCL-1/BH3蛋白相互作用的α-螺旋模拟化合物,本发明人在聚合物珠上构建了具有三嗪-哌嗪-三嗪作为核心结构的化学库,并且根据图4中的珠上筛选法(on-bead screening method)选择直接与MCL蛋白结合的化合物。
结果证实,化学式4至6的化合物与MCL-1结合并且抑制MCL-1与BH3蛋白之间的相互作用,并且发现这些化合物诱导癌细胞凋亡(图6至图10)。此外,在本发明中,将多种亲水基团引入到化学式6化合物的R4和R5位置以获得大量的具有增强的对MCL-1的结合能力的衍生物(图11和图12)。
为了证明三嗪-哌嗪-三嗪化合物可充当用于抑制蛋白质间相互作用的α-螺旋模拟物,从本发明构建的库中选择化学式10的化合物作为抑制BH3蛋白与MCL-1蛋白之间的相互作用的化合物。发现化学式10的化合物与MCL-1结合并且抑制MCL-1与BH3蛋白之间的相互作用,证实这些化合物诱导癌细胞凋亡(图16和图17)。
因此,化学式4至6和化学式9、10的化合物可用作癌症的预防或治疗剂。
在另一个方面,本发明涉及用于预防和治疗帕金森病的药物组合物,其包含选自以下化学式7和8化合物的至少一种化合物或其可药用盐。在另一个实施方案中,本发明涉及用于治疗帕金森病的方法,其包括向个体施用有效量的选自化学式7和8的化合物的至少一种化合物或其可药用盐:
[化学式7]
[化学式8]
α-突触核蛋白的聚集诱导路易体(Lewy bodies)的形成,其被认为是帕金森病的病理特征。因此,为了提供本发明中的α-螺旋模拟化合物可通过稳定α-突触核蛋白的折叠结构抑制α-突触核蛋白聚集,抑制聚集的化合物选自本发明构建的化学库。结果证实,化学式7至8的化合物与α-突触核蛋白结合并抑制α-突触核蛋白的聚集(图13和图14)。
因此,化学式7和8的化合物可用作用于帕金森病的预防或治疗剂。
组合物还可包含常规用于生产药物组合物的合适的载体、赋形剂和稀释剂。组合物可配制成片剂、胶囊剂、胶囊剂、混悬剂、乳剂、糖浆剂、外用制剂、栓剂、无菌注射溶液等。
当配制组合物时,通常使用稀释剂或赋形剂,例如填充剂、增量剂、黏合剂、润湿剂、崩解剂、表面活化剂等。用于经口施用的固体制剂包括片剂、丸剂、散剂、颗粒剂、胶囊剂等,并且这样的固体制剂可包含至少一种赋形剂和/或润滑剂。用于经口施用的液体制剂包括混悬剂、溶液剂、乳剂和糖浆剂。除了水和液体石蜡(其是常用的稀释剂)之外,还可包括多种赋形剂,例如润湿剂、甜味剂、芳料、防腐剂等。用于肠胃外施用的制剂可包括无菌水溶液、非水溶剂、混悬剂、乳剂、冻干制剂、栓剂等。
组合物的优选剂量根据患者的状况和体重、疾病的程度、药物形式、施用途径和周期等而变化,但是可由本领域技术人员适当地选择。为了更优选的效果,本发明的组合物的剂量基于活性成分优选地为每天0.1mg/kg至20mg/kg,但不限于此。施用可每天进行一次或分成数次。本发明的组合物可以多种方式施用于哺乳动物(包括人)。可以预期所有施用方式,例如通过经口、静脉内、肌内、皮下注射等。本发明的组合物的药物剂型可以以活性成分的可药用盐的形式使用,并且可以单独使用或与其他药物活性化合物组合使用以及以合适的组合使用。
[实施例]
在下文中,将参照实施例对本发明进行详细地描述。然而,以下实施例用于举例说明本发明,并且本发明不受以下实施例的限制。
实施例1.包含三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的制备
1-1.实验材料的制备
TentaGel B NH2/Boc树脂(130μm,0.25mmol/g)和TentaGel S NH2树脂(130μm,0.29mmol/g)购自Rapp Polymere。Rink amide MBHA树脂(0.96mmol/g)购自Novabiochem。在Agilent 1200 LC/MS系统(Agilent Technology)上在C18反相柱(Kinetex,2μm,4.6mm*50mm)上进行液相色谱/质谱(LC-MS)分析。通过使用溶剂A以90%梯度洗脱2分钟并通过使用溶剂B以100%梯度洗脱14分钟来进行LC-MS,流量为0.8mL/分钟(包含95%水、5%甲醇和0.01%TFA的溶剂A;以及包含甲醇和0.01%TFA的溶剂B)。在Agilent 1120 Compact LC系统(Agilent Technology)上使用C18反相柱(Agilent Technology,5μm,25mm*125mm)进行HPLC纯化。使用从10%B溶剂至100%B溶剂的线性梯度,同时每40分钟改变溶剂组成。使用ABI 4800和ABI 5800质谱仪(Applied Biosystems)进行MALDI-TOF MS并使用α-氰基-4-羟基肉桂酸作为底物。
1-2.使用固相合成法的α-螺旋模拟化合物的制备
为了合成基于三嗪-哌嗪-三嗪的化合物,开发了固相合成法,如图2中所示。通过使用经胺(NH2)修饰的聚合物珠通过五个顺序偶联反应以92%的平均收率获得期望的化合物。
参照图2,作为第一步,将单取代的二氯三嗪(“3”)加载到Rink-Amide MBHA树脂上并与树脂结合以产生与树脂结合的三嗪衍生物“4”。在第二步中,通过将用2-硝基苯磺酰基(Ns)保护的哌嗪衍生物(“5”)与与树脂连接的三嗪衍生物(“4”)偶联来制备化合物“6”。在第三步中,从上述偶联的化合物(“6”)中除去N-Ns保护基,然后添加2-乙基氢基-4,6-二氯-[1,3,5]三嗪而将其引入以产生化合物“8”。在第四步中,用多种胺基团(R2”R2“NH)取代上述制备的化合物(“8”)的氯位点。在第五步中,添加三氟乙酸(TFA),制备用三个取代基官能化的化合物“9”。
如下将对制备过程进行更详细地描述。
将Rink-Amide MBHA树脂(100mg,69μmol)在5mL烧结注射器中在DMF(2mL)中处理1小时并用DMF中的20%哌啶处理(2×10分钟)以除去Fmoc基团。在用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)洗涤后。为了将具有单取代的二氯三嗪的树脂加载到树脂上,添加DMF(1mL)并与Et3N(5当量)在室温下反应过夜。为了将用2-硝基苯磺酰基(Ns)保护的哌嗪衍生物(“5”)(10当量)与树脂偶联,将产物与Et3N(5当量)在DMF中于60℃下孵育过夜。除非另有说明,否则在每个反应步骤结束时用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤树脂。为了从偶联的化合物(“6”)中除去N-Ns保护基,使化合物与2-巯基乙醇(20当量)和DBU(10当量)在DMF中于室温下反应2小时。然后为了通过引入2-乙基氨基-4,6-二氯-[1,3,5]三嗪(“7”)来制备化合物(“8”),用化合物7(5当量)和NMP中的DIEA(5)处理珠并使其在60℃下反应过夜。最后,添加TFA(三氟乙酸)以进行切割反应,以产生用三个取代基官能化的哌嗪-三嗪化合物“9”。
用LC-MS分析由固相合成法制备的化合物的纯度和鉴定。结果,最终产物的平均纯度为92%或更高,证实了固相合成法的效力。用反相HPLC纯化化合物并用1H-NMR、13C-NMR和HRMS(高分辨率质谱仪)进行鉴定。
[表2]
代表性的40种最终产物的质量和纯度数据。
实施例2.包含具有三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟物的化学库的构建
在本发明中,使用用胺(NH2)修饰的聚合物珠构建包含三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟物化学库,并且将其命名为OBOC(一珠一化合物,one-bead one-compound)库。制备过程在图3中示出。
如图3所示,该实施例使用双官能的珠,其中珠的表面(外部)侧结合用BOC(叔丁氧羰基)基团保护的胺基团且珠的内侧结合游离胺。在珠的表面上,根据实施例1中描述的固相合成法合成α-螺旋模拟物,并且在每次将取代基引入到α-螺旋模拟物中时,将对应于每个取代基的类肽残基与珠的内侧连接,以最终获得与一个珠的两种化合物连接。换句话说,α-螺旋模拟物在珠的表面上而对α-螺旋模拟物呈特异性的类肽在珠的内侧上。该类肽充当编码标签,其允许α-螺旋模拟物的结构与珠表面结合。在一些情况下,对应于α-螺旋模拟物的各个取代基的类肽的取代基可以相同。然而,当α-螺旋模拟物的取代基为仲胺或哌嗪时,类肽的取代基可与α-螺旋模拟物的取代基不同。因此,在本发明中,通过分别标记类肽的R5取代基上的18种、R3取代基上的3种和R2取代基上的27种来制备α-螺旋模拟化合物的多种组合。结果,可构建包括总计1,458种(=18×3×27)的化学库。
[表3]
与用于库合成的类肽标签偶联的取代基的结构。括号中结构表示当取代基不同时用于编码类肽合成的胺。
构建的α-螺旋模拟物的化学库可用于珠上的高效筛选。过程在图4中示出。
如图4所示,在珠筛选中,珠表面上的α-螺旋暴露于靶蛋白,但是珠内部的类肽未暴露。筛选之后,可通过对珠内部的类肽进行测序来分析珠表面上的α-螺旋模拟物的结构。
如下将对制造过程进行更详细地描述。
用DMF(2mL)处理TentaGel B NH2/Boc树脂(250mg,NH2:62.5μmol/Boc:1.3μmol)2小时。在DMF(1mL)中的HOBt(5当量)、HBTU(5当量)和DIEA(10当量)的存在下,用Fmoc-L-甲硫氨酸(Fmoc-Met-OH)(5当量)处理珠。在搅拌2小时之后,移除反应混合物并用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)进行洗涤。接着,使用相同的类肽偶联条件添加Fmoc-4-氨基丁酸(Abu)(5当量)。重复以上过程两次以偶联三个Abu接头残基。在Fmoc去保护之后,通过在室温下用DMF(2mL)中的1M溴乙酸和1M DIC进行处理而使所得的胺溴乙酰化。为了用烯丙氧羰基(Alloc)基团保护类肽的N-末端,对无水CH2Cl2中的氯甲酸烯丙酯(5当量)和Et3N(6当量)进行处理并且使其在4℃下反应过夜。为了除去珠表面上的Boc基团,添加CH2Cl2中的50%TFA并反应1小时。使所得伯胺与18种另外的单取代的三嗪衍生物(“4”)(5当量)偶联。在用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤之后,将所有的珠随机分入250mL容器中并分成3个容器。对DIEA(10当量)中的Boc保护的哌嗪(10当量)和NMP(1mL)进行处理并在60℃下反应过夜以用三种不同的哌嗪取代三嗪的氯位置。在用DMF(3×)、MeOH(2×)、CH2Cl2(2×)洗涤之后,对无水CH2Cl2(1mL)中的Pd(PPh3)4(0.2当量)和PhSiH3(10当量)进行处理以除去Alloc基团。通过使用亚单体途径偶联第二类肽残基(R2”)。将珠再次混合并使其随机化。在溴乙酰化之后,将珠分成27个容器并与27种不同的胺反应。通过溴乙酰化和随后与哌啶的反应来封闭类肽的N-末端。接着,对CH2Cl2中的50%TFA进行处理并反应1小时以除去Boc基团。在用DMF中的10%DIEA中和之后,向珠中添加NMP中的DIEA和4,6-二氯-1,3,5-三嗪-2-乙胺并在60℃下反应过夜。在用DMF(3×)、MeOH(2×)和CH2Cl2(2×)洗涤之后,将所有的珠混合并通过用95%TFA处理来进行总体去保护。为了切割,对于每个珠用ACN/AcOH/H2O(5∶4∶1)中的20μL CNBr(40mg/mL)进行处理并在室温下反应12小时。
为了证实库合成的效力,从库中随机选择30个珠。在用CNBr进行切割反应之后,通过MS分析所得的类肽。通过CNBr处理,珠表面上的α-螺旋样分子未被切割,而是仅释放通过甲硫氨酸与珠连接的类肽。在所有测试的珠中未观察到显著的杂质峰,证实了库合成的高纯化率和效力。
实施例3.抑制BH3(BCL-2同源结构域3)蛋白与MCL-1(髓样细胞白血病-1)蛋白之间的相互作用的α-螺旋模拟化合物的选择和活性测试
3-1.化合物选择
在本发明中,为了证明三嗪-哌嗪-三嗪化合物可充当抑制蛋白质间相互作用的α-螺旋模拟物,从在实施例2中构建的库中选择对BH3蛋白与MCL-1蛋白之间的相互作用具有抑制活性的化合物。
MCL-1(髓样细胞白血病-1)是具有抗凋亡活性的BCL-2(B细胞淋巴瘤2)家族蛋白之一,并且抑制由BH3引起的细胞凋亡并通过与BH3(BCL-2同源结构域3)家族蛋白(例如BAK)相互作用来帮助细胞存活。因此,抑制癌细胞中MCL-1蛋白与BH3蛋白之间的相互作用可防止癌细胞存活,从而是有前景的抗癌治疗策略。
为了选择抑制MCL-1/BH3蛋白相互作用的α-螺旋模拟化合物,本发明人根据实施例2的相同方法在聚合物珠上构建了化学库,并且通过使用如图4所示的珠上筛选法选择与MCL-1蛋白直接结合的化合物。第一,将25mg(约0,200珠,~多样性拷贝)的包含与珠25mg(约0,200珠,~多样性拷贝)结合的化合物的库与200nM的生物素化的MCL-1ΔNΔC(生物素化的MCL-1)在4℃下孵育过夜。洗涤未结合的蛋白质并用与链霉亲和素偶联的Dynabead(铁氧化物颗粒)进行处理,以使用磁性分离来分离阳性珠。在第一筛选步骤中选择数百个珠。在用MCL-1进行筛选之前,除去与链霉亲和素缀合的珠(潜在的假阳性)。在1%SDS下加热所选择的珠以除去结合的蛋白质。在洗涤之后,将珠与100nM的生物素化MCL-1ΔNΔC在4℃下再孵育4小时并用链酶亲和素缀合的碱性磷酸酶进行标记。为了选择最佳的化合物,在严格条件(即,更少量的蛋白质和更短的孵育时间)下进行二次筛选。接着,用5-溴-4-氯-3-吲哚基磷酸酯处理珠用于碱性磷酸酶的比色检测。分离出十个蓝珠作为候选化合物。用MS/MS分析来分析来自每个珠的类肽。结果,10个珠包含相同的类肽序列并且可将类肽主要分成3种类肽组。5个珠包含类肽-1,3个珠包含类肽-2,以及2个珠包含类肽-3。
[表4]
该结果被认为是由于库中包括每种化合物的约7个拷贝,表明本发明中开发的筛选系统工作良好。作为分析三种类肽的结果,所选择的化合物分别具有以下9a(化学式4)、9b(化学式5)和9c(化学式6)的结构。通过HPLC分离这些化合物并发现其是水溶性的(这些化合物以约100μg/mL溶解在pH 7.4的磷酸盐缓冲盐水中,表明在药物使用中待用作口服吸收的水溶解度的范围)。
[化学式4]
[化学式5]
[化学式6]
3-2.荧光标记化合物的合成
用于荧光标记化学式4至6的化合物的过程在图5中示出,并且荧光标记化合物分别显示为9a-FL、9b-FL和9c-FL。
第一,用DMF(2mL)处理TentaGel S NH2树脂(100mg,29μmol)1小时。在DMF(1mL)中的HOBt(5当量)、HBTU(5当量)和DIEA(10当量)的存在下,用Fmoc-L-甲硫氨酸(Fmoc-Met-OH)(5当量)处理珠并在室温下反应。在搅拌2小时之后,得到反应混合物并洗涤。为了除去Fmoc保护基,用DMF(1mL,2×10分钟)中的20%哌啶处理所得的产物。接着,在相同的肽偶联条件下将Fmoc-Lys(Alloc)-OH与Fmoc-Abu-OH偶联。在Fmoc去保护之后,通过DMF中的DIEA(5当量)和单取代的二氯三嗪的处理,使获得的产物在室温下反应过夜。得到反应混合物并用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和NMP(3×)洗涤。将Boc保护的哌嗪衍生物(10当量)和DIEA(10当量)添加到NMP(1mL)中的珠中。将混合物在60℃下搅拌过夜。在洗涤之后,将CH2Cl2中的50%TFA处理1小时以除去Boc基团。将树脂用DMF(1mL)中的10%DIEA中和1小时,然后用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和NMP(3×)洗涤。然后,将NMP中的DIEA(5当量)和4,6-二氯-1,3,5-三嗪-2-乙胺添加至珠中,在60℃下反应过夜并用DMF(3×)、MeOH(2×)和NMP(3×)洗涤。然后用NMP中的DIEA(20当量)和胺(20当量)处理树脂,在60℃下反应过夜并用DMF(3×)、MeOH(2×)和CH2Cl2(2×)洗涤。通过在无水CH2Cl2(1mL)中处理Pd(PPh3)4(0.2当量)和PhSiH3(10当量)1小时来去保护Alloc之后,通过使用肽偶联条件使产生的赖氨酸残基的NH2与5,6-羧基荧光素(5当量)偶联。为了切割,用ACN/AcOH/H2O(5∶4∶1)中的20μL CNBr(40mg/mL)处理珠并在室温下反应12小时。通过HPLC纯化切割粗制产物。
3-3.计算机辅助分子对接研究
使用Maestro(Maestro v9.0,Schrdinger,LLC,Portland,OR)构建化合物的结构,并通过使用CHARMM Gen-eral Force Field(Vanommeslaeghe,K;Hatcher,E.;Acharya,C.;Kundu,S.;Zhong,S.;Shim,J.;Darian,E.;Guvench,O.;Lopes,P.;Vorobyov,I.;Mackerell,A.D.J.Comput.Chem.2010,31,671)获得最低能量。通过使用Raccoon(Forli,S.Raccoon utodock VS:an automated tool for preparing AutoDock virtual screenings)分析用于对接的配体和受体的结构。对接盒中心组织有BH3螺旋残基中的几何中心并且盒尺寸的X、Y和Z设定为25。为了在MCL-1晶体结构中产生位姿诱饵(pose decoy)(PDB条目:3MK8),进行了对每种化合物的独立的20AutoDock Vina(Trott,O.;Olson,A.J.J.Comput.Chem.2010,31,455)对接。对于每种化合物得到了400种位姿诱饵并且最终模型确定为最佳得分位姿。用软件PyMol显现化合物的分子结构。
3-4.蛋白质纯化
用GST标记人MCL-1ΔNΔC蛋白中编码BH3-结合结构域(第172至320位氨基酸)的表达载体并将其引入BL21(DE3)大肠杆菌(E.coli)细胞中。用1mM IPTG(异丙基β-D-1-硫代吡喃半乳糖苷)处理细胞以诱导蛋白质表达。用裂解缓冲液(20mM Tris,pH 7.2,250mM NaCl,完全蛋白酶片(Roche))使细胞沉淀重悬并进行超声处理以裂解。在离心之后,将细胞裂解物施加于5mL GSTrap HP柱(GE Life Sciences)并根据造商的说明书进行操作。MCL-1蛋白通过如下获得:用凝血酶蛋白酶切割,重新加载到GSTrap HP柱上并通过尺寸排阻色谱以除去GST。将MCL-1蛋白与磺基-NHS-生物素(Thermo Scientific)在0.1M NaHCO3缓冲液(pH 8.3)中于室温下孵育45分钟以使MCL-1蛋白生物素化(因为测试中使用的MCL-1ΔNΔC蛋白具有若干个暴露在表面上并且远离BH3结合的活性位点的赖氨酸残基,所以蛋白质通过修饰而被生物素化)。通过添加1.5M羟胺使反应淬灭。通过透析用包含0.05%吐温20(pH 7.4)的1×PBS缓冲液分离经标记的蛋白质。根据制造商的说明书确定标记的程度(生物素化的程度:每个蛋白质1.05个生物素)。
3-5.荧光偏振测定
使用在黑色Costar 384孔板中调整至60μL的最终浓度的结合缓冲液(50mM Tris,100mM NaCl,pH 8.0),将荧光标记的化合物(50nM)与MCL-1ΔNΔC或BCL-1在黑暗中于室温下孵育30分钟。用SpectraMax M5多模式微板读数器(Molecular Devices)测量荧光偏振值。激发波长调整至485nm并且在535nm处测量发射。用GraphPad Prism 4软件计算KD。为了进行竞争性荧光偏振测定,用FITC标记的MCL-1 BH3肽(FITC-MCL-1-BH3肽)替代MCL-1ΔNΔC。合成N-末端标记的FITC-MCL-1-BH3肽(FITC-Abu-KALETLRRVGDGVQRNHETAF-NH2)并用HPLC纯化。将不同浓度的化合物与FITC-MCL-1-BH3(50nM)和0.8μM MCL-1ΔNΔC在结合缓冲液中孵育30分钟,然后添加FITC-MCL-1-BH3(50nM)。在添加不同浓度的化合物进行孵育之后,测量荧光偏振。使用常规方法计算Ki(Nikolovska-Coleska,Z.等,Anal.Biochem.2004,332,261)。
3-6.细胞生存力测定
在包含10%FBS的RPMI 1640培养基中培养Jurkat T细胞,并将4×104个Jurkat T细胞接种在96孔板的100μL Opti-MEM培养基(Invitrogen)中。在包含10%FBS的MEM培养基中培养WS-1细胞并将2×104个WS-1细胞接种在96孔板的包含10%FBS的100μL MEM培养基中24小时,用PBS洗涤,并在100μL Opti-MEM中进行培养。将细胞维持在血清饥饿状态下6小时并用化合物进行处理。在培养24小时之后,使用CellTiter 96AQueous非放射性细胞增殖测定试剂盒(Promega)根据制造商的说明书测定细胞生存力。
3-7.胱天蛋白酶测定
将Jurkat T细胞以2×104个细胞/孔平板接种在白壁96孔板的Opti-MEM培养基中并用不同浓度的化合物或DMSO处理16小时。将WS-1细胞以1×104个细胞/孔平板接种在白壁96孔板上并在包含10%FBS的MEM培养基中培养24小时。用PBS洗涤细胞并用Opti-MEM培养基中的化合物进行处理,如Jurkat T细胞。使用胱天蛋白酶-Glo 3/7测定试剂盒(Promega)根据制造商的说明书测量胱天蛋白酶3/7活性。
3-8.共聚焦显微术实验
将A549细胞(5×103个细胞)平板接种在96孔板的包含10%FBS的MEM培养基中。将细胞保持24小时,用PBS洗涤,然后在Opti-MEM培养基中处理1μL荧光标记的化合物或5(6)-羧基荧光素并孵育2.5小时。用1×PBS洗涤细胞(2次)并分析荧光图像。
3-9.免疫共沉淀测定
在Opti-MEM培养基中,用DMSO或化合物处理Jurkat T细胞(1×107个细胞)。在培养2.5小时之后收获细胞,用PBS洗涤一次,并使用裂解缓冲液(50mM Tris-HCl,pH 7.4,150mM NaCl,1mM EDTA,1mM DTT,0.5%NP-40和完全蛋白酶抑制剂片)在冰上裂解。将细胞裂解物与抗MCL-1抗体(S-19,Santa Cruz)在4℃下孵育过夜并用蛋白A/G琼脂糖孵育1小时。使珠沉淀,用裂解缓冲液洗涤三次,并使用抗BAK(Cell Signaling)和抗MCL-1抗体对变性蛋白质进行蛋白质印迹。在Immobilon-FL(Millipore)PVDF膜上进行蛋白质印迹分析。使用山羊抗兔IRDye 680抗体(Licor)作为二抗并用Odyssey 3.0软件分析信号。
3-10.实验结果
作为通过化合物的荧光偏振测定测量MCL-1蛋白与所选择的三种化合物的结合亲和力的结果,化学式4至6的化合物与MCL-1结合的KD值分别为2.8、3.0和1.1μM,证实所有三种化合物均有效地与MCL-1蛋白结合。用作阳性对照的已知BH3肽的KD值为0.3μM。用作阴性对照的氟接头不与MCL-1结合,表明与每种化合物结合的荧光团和接头不影响与MCL-1结合(图6B)。此外,进行竞争性荧光偏振测定以便这些化合物实际上干扰MCL-1与BH3蛋白之间的相互作用。结果,解离MCL-1与BH-3之间的相互作用的化学式5和6的化合物的解离Ki值分别测量为17.2μM和9.3μM,并且化学式4化合物表现出弱的活性。用作阴性对照的化合物9-nc不与MCL-1结合(图6C)。接着,基于计算机的对接模型研究预测化学式6的化合物将很好地与MCL-1的BH3结合部位结合(图7B)。
BH3变得游离并引起癌细胞凋亡。因此,通过与MCL-1组合抑制MCL-1与BH3蛋白之间的相互作用,研究所选择的化合物是否诱导癌细胞死亡。
第一,为了证实化学式5的化合物抑制细胞中MCL-1/BH3蛋白相互作用,用化学式6的化合物处理Jurkat白血病T细胞(癌细胞)。通过抗MCL-1抗体的处理使细胞裂解物免疫沉淀,然后使用抗BAK和抗MCL-1抗体进行蛋白质印迹,证实发现化学式5的化合物有效抑制MCL-1和BAK(BH3蛋白之一)(图8A)。然而,阴性对照化合物9nc不抑制MCL-1与BAK之间的相互作用(图9)。此外,发现化学式6的化合物渗入细胞中并竞争性地与MCL-1结合和释放BAK蛋白。共聚焦显微术分析表明化合物对细胞是可渗透的(图8B)。化学式6的化合物有效地抑制Jurkat白血病T细胞和多发性骨髓瘤细胞U266细胞的癌细胞的存活,但是不影响WS-1人成纤维细胞和HEK293胚胎肾细胞的正常细胞的存活。因此,证实化学式6的化合物对癌细胞具有选择性作用(图8C和图10B、C)。
作为用于证实由化学式6的化合物的细胞凋亡引起的癌细胞死亡的胱天蛋白酶3/7测定的结果,化学式6的化合物在U266细胞中以浓度依赖性方式提高胱天蛋白酶活性,但在WS-1细胞中不提高(图10C)。
结果,化学式4至6的化合物抑制BH3蛋白与MCL-1蛋白之间的相互作用。其中,化学式6的化合物选择性地在癌细胞中表现出细胞毒性。因此,这些化合物在未来作为抗癌药物是非常有前景的。此外,证实了本发明中开发的三嗪-哌嗪-三嗪支架化合物充当α-螺旋模拟物并且可有效地抑制蛋白质-蛋白质相互作用。
实施例4.制备包含三嗪-哌嗪-三嗪作为核心结构的α-螺旋模拟化合物的具有改善的亲水性的衍生物
在对BH3蛋白与MCL-1蛋白之间的相互作用表现出优异的抑制作用的化学式6的化合物中,R1位处的Et和R4位置的H被多种亲水性官能团替代,以开发具有比化学式6的化合物对MCL-1更高的结合亲和力的衍生物。
在化学式6的化合物中,R1位置的Et被多种亲水性官能团替代以得到10种衍生物,称为MO-01至MO-10,并且制备过程在图11中示出。
在化学式6的化合物中,R4位置的H被多种亲水性官能团取代以得到10种衍生物,称为MO-11至MO-20,并且制备过程在图12中示出。
制备的MO-01至MO-20的衍生物如下所示:
[表5]
实施例5.对α-突触核蛋白聚集具有抑制作用的α-螺旋模拟化合物的选择和效力测试
5-1.化合物选择
为了证明包含三嗪-哌嗪-三嗪的化合物可使蛋白质折叠稳定并抑制蛋白质聚集,使用α-突触核蛋白作为与蛋白质折叠相关的代表性疾病因子。α-突触核蛋白的聚集诱导路易小体的形成,这被认为是帕金森病的病理特征。尽管路易小体的形成机制尚不清楚,但认为α-突触核蛋白的错误折叠及其不适当的自缔合参与路易小体的形成。还已知,约65%的α-突触核蛋白由α-螺旋构成。因此,已经实验性地检查了本发明中开发的α-螺旋模拟化合物是否可使α-突触核蛋白的折叠稳定并抑制α-突触核蛋白聚集。
为了实验的目的,通过在α-突触核蛋白的N末端引入MKCK四肽(即,由甲硫氨酸-赖氨酸-半胱氨酸-赖氨酸构成的肽)以及能够与荧光团和珠化学交联的反应性半胱氨酸(野生型α-突触核蛋白不包含半胱氨酸)来制备构建物。在使包含半胱氨酸的α-突触核蛋白(cys-Syn)生物素化之后,根据实施例3-1中描述的方法筛选化合物。结果,选择化学式7(Q1)和化学式8(Q2)的化合物。
[化学式7]
[化学式8]
5-2.蛋白质纯化
从pRS载体,扩增N末端延伸MKCK四肽的开放阅读框和α-突触核蛋白的构建物。使用包含XhoI限制性位点的反向引物(5′-GCCTTTGAAAGTCCTTTGCAGAATACACACATTCCCGGGAATTCC-3′)和包含SamI限制性位点的正向引物(5′-GGAATTCCCGGGAATGTGTGTATTCTGCAAAGGACTTTCAAAGGC-3′)。将扩增的DNA克隆到pGEX-6P-1质粒(GE Biosciences)中以产生GST融合构建物。将表达质粒引入rossetta 2(DE3)大肠杆菌细胞中并通过在20℃下用1mM IPTG处理来诱导蛋白质表达。使细胞沉淀重悬于裂解缓冲液(100mM HEPES,pH=7.4,150mM NaCl,10%甘油,0.1%BOG,Halt蛋白酶抑制剂混合物(Thermo Scientific))中并进行超声处理以裂解。在离心之后,将细胞裂解物施加于20mL GSTrap HP柱(GE Life Sciences)并根据造商的说明书进行操作。α-突触核蛋白蛋白质通过与PreScission蛋白酶(GE healthcare life sciences)的切割反应过夜获得。在缓冲液更换之后,再次加载GSTPrep柱并进行尺寸排阻色谱以除去GST。
为了使α-突触核蛋白蛋白质生物素化,将α-突触核蛋白与磺基-NHS-生物素(Thermo Scientific)在0.1M NaHCO3缓冲液(pH 8.3)中于室温下培养45分钟。通过添加1.5M羟胺使反应淬灭。使用包含0.05%吐温20的1×PBS缓冲液(pH 7.4)通过透析分离标记的蛋白质并根据制造商的说明书确定标记程度(生物素化的程度:每个蛋白质1.07个生物素)。
5-3.硫磺素T聚集测定
将每种化合物和2-10uL DMSO(作为空白)用100uL 0.2mMα-突触核蛋白疾病相关的突变体A53T细胞处理并用缓冲液(100mM HEPES,pH 7.4,150mM NaCl,10%甘油,0.1%BOG和5uM硫磺素T)孵育以得到100μM的最终浓度并在频繁搅拌下于37℃下孵育。使用FlexStation 2(Molecular Devices,Sunnyvale,CA,USA)在37℃下以1小时的间隔测量硫磺素T荧光(激发波长440um、发射波长485nm、截止波长475nm),总时间为30小时。
5-4.圆二色性(CD)热变性测定
在存在或不存在化合物的情况下,在配备有Peltier温度控制器的BioLogic MOS-450分光光度计上使用1cm路径长比色皿用222nm CD信号监测α-突触核蛋白的热变性。在25℃至60℃和1℃/分钟下向蛋白质样品(0.05mg/mL,100mL HEPES,150mM NaCl,10%甘油,0.1%BOG,1%牛磺脱氧胆酸)中添加DMSO溶液和化合物以得到1%DMSO的1uM最终浓度。
5-5.实验结果
为了确定所选择的化学式7和8的化合物是否与α-突触核蛋白结合,如实施例3至5中所述测量α-突触核蛋白的KD值。结果,化学式7和8的化合物KD值分别测量为68nM和148nM。这两种化合物均有效地与α-突触核蛋白结合(图13B)。化学式7和化学式8的化合物在结构上相似,因为R3和R5具有相同的取代基且R2为1,2-二烷氧基苯(图13A)。
为了找出化合物支架和荧光团接头在化学式7和8的化合物与α-突触核蛋白结合中如何表现,测试了具有与化学式7和8相同的支架的化学式4至6(9a至9c)的化合物以及氟接头与α-突触核蛋白结合的结合程度。结果,与化学式7和8的化合物具有两个相同的取代基的化学式6的化合物表现出对α-突触核蛋白的亲和力(图13B),支持了化合物对靶蛋白的结合和选择性由化合物的侧链决定。
为了测试化学式7和8与α-突触核蛋白的结合是否影响α-突触核蛋白的热稳定性,进行圆二色性(CD)热变性测定。结果,化学式7和8的化合物将α-突触核蛋白的解链温度从37℃分别改变为40℃、41℃。已知α-突触核蛋白的α螺旋具有长的疏水表面和两亲性质,化学式7和8的相对疏水的化合物与α-突触核蛋白的疏水表面相互作用。
为了测试化学式7和8的化合物是否影响α-突触核蛋白的聚集,使用α-突触核蛋白疾病相关的突变体A53T细胞。第一,测量突变体(A53T)的KD值的结果,化学式7和8的化合物的KD分别为268nM和372nM,证实了这两种化合物均有效地与突变体α-突触核蛋白结合(图14A)。将化学式7和8的每种化合物和化学式4的化合物(9a)与突变体α-突触核蛋白孵育并根据硫磺素T聚集监测聚集。在实验的30小时期间,化学式7和8的化合物显著地延迟了α-突触核蛋白聚集的发生并且显著地降低了聚集的原纤维的量,而用化学式4的化合物和用作阴性对照的DMSO处理的组不抑制聚集(图14B)。
结果,证实了本发明的α-螺旋模拟库的化合物与α-突触核蛋白结合并且可抑制α-突触核蛋白的聚集。因此,根据本发明的用多种取代基取代的三嗪-哌嗪-三嗪支架化合物的化学库不仅可用于MCL-1或α-突触核蛋白而且可用于能够控制通常通过α-螺旋介导的蛋白质相互作用的抑制剂。
实施例6.包含苯基-哌嗪-三嗪作为核心结构的化合物的制备
为了合成基于苯基-哌嗪-三嗪的化合物,如图15所示开发固相合成法。通过用胺(NH2)修饰的珠的顺序偶联反应以90%或更高的平均收率获得期望的化合物。
参照图15,作为第一步,加载4-氟-3-硝基苯甲酸并使其与Rink-Amide MBHA树脂结合,得到与树脂结合的化合物(“2”)。在第二步中,通过将用N-硝基苯磺酰基(Ns)保护的哌嗪衍生物(“3”)与化合物(“2”)偶联来制备化合物(“4”)。在第三步中,通过用2-巯基乙醇处理从偶联的化合物(“4”)中除去N-Ns保护基,随后添加2-乙基氨基-4,6-二氯-[1,3,5]三嗪(“5”)而将其引入以产生化合物(“6”)。在第四步中,通过用多种胺基团(R3NH2)替代化合物(“6”)的氯位置来制备化合物(“7”)。在第五步中,通过使SnCl2在DMF中反应以还原化合物的硝基来制备化合物(“8”)。通过使用多种醛的还原胺化反应将所得的胺烷基化以引入R1官能团。与芳香醛的反应以高收率产生期望的烷基化产物,但是在相同条件下与脂肪醛的反应产生作为主要产物的二烷基化产物。为了防止由于过度烷基化而形成副产物,向反应混合物中添加苯并三唑。已知苯并三唑在还原胺化中抑制二烷基化副产物的形成。作为最后一步,用95%TFA进行切割反应以产生用三个取代基官能化的苯基-哌嗪-三嗪衍生物。
如下将对制备过程进行更详细地描述。
用DMF(2mL)处理Rink-Amide MBHA树脂(100mg,93μmol)1小时并用DMF中的20%哌啶处理(2×10分钟)以除去Fmoc基团。除非另有说明,否则在每个反应步骤结束时用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤树脂。将树脂添加至4-氟-3-硝基苯甲酸(70mg,378μmol)、HATU(142mg,378μmol)和DIEA(130μL,756μmol)在DMF(1.5mL)中的溶液中并使混合物在室温下反应12小时。为了向树脂中引入硝基苯磺酰基保护的哌嗪(4当量),使树脂与NMEA中的DIEA(10当量)在95℃下反应12小时以获得与树脂结合的哌嗪衍生物“4”。
用DMF中的2-巯基乙醇(20当量)和DBU(10当量)处理产物并在室温下反应以除去衍生物“4”的硝基苯磺酰基。然后,为了将2-乙基氨基-4,6-二氯-[1,3,5]三嗪(10当量)与树脂偶联,使THF中的DIEA(5当量)在60℃下反应3小时以产生化合物“6”。为了用多种胺基团(R3NH2,5当量)替代化合物“6”的氯,使产物在NMP中的DIEA(10当量)的存在下在80℃下反应过夜以获得化合物“7”。将DMF中的SnCl2·2H2O(60当量)添加至树脂中并将混合物在室温下搅拌24小时。用混合溶液(CH(OMe)3∶DMF=9∶1)中的0.4M醛溶液(10当量)和甲醇中的20%(v/v)乙酸处理与树脂结合的胺(“8”)并在50℃下反应18小时。然后,添加TMF中的5M NaCNBH3(50当量)并将混合物在50℃下搅拌6小时以产生烷基化产物。当使用脂肪醛时,添加苯并三唑(100当量)以防止二烷基化副产物的形成。最后,用TFA进行切割以产生用三个取代基官能化的苯基-哌嗪-三嗪衍生物。
用LC-MS分析由固相合成法制备的化合物的纯度和鉴定。结果,最终产物的平均纯度为90%或更高,证实了固相合成法的效力。用反相HPLC纯化化合物并用1H-NMR、13C-NMR和HRMS(高分辨质谱仪)进行鉴定。
36种制备的α-螺旋模拟物示于表6中。
[表6]
实施例7.抑制BH3蛋白与MCL-1蛋白之间相互作用的α-螺旋模拟化合物的选择和活性测试
7-1.蛋白质纯化
用GST标记编码人Mcl-1172-320(第172至320位氨基酸)或人Bcl-xL1-212(第1至212位氨基酸)中的BH3结合结构域(第172至320位氨基酸)的表达载体并将其引入BL21(DE3)大肠杆菌细胞中。用1mM IPTG(异丙基β-D-1-硫代吡喃半乳糖苷)处理细胞以诱导蛋白质表达。用裂解缓冲液(20mM Tris,pH 7.2,250mM NaCl,完全蛋白酶片(Roche))使细胞沉淀重悬并进行超声处理以裂解。在离心之后,将细胞裂解物施加于5mL GSTrap HP柱(GE Life Sciences)并根据造商的说明书进行操作。通过用凝血酶蛋白酶切割来获得Mcl-1172-320和Bcl-xL1-212蛋白,重新加载到GSTrap HP柱上并通过进行尺寸排阻色谱以除去GST。
7-2.荧光偏振测定
为了进行竞争性荧光偏振测定,用荧光标记的BAK-BH3肽(FITC-BAK-BH3肽)替代MCL-1172-320。具体地,将10nm的TAMRA标记的Bak-BH3(TAMRA-Abu-KALETLRRVGDGVQRNHETAF-NH2)肽与0.8μM的Mcl-1172-320在黑色Costar 384孔板中的最终体积为60μL的结合缓冲液(50mM Tris,100mM NaCl,20nM牛血清白蛋白,pH 8.0)中在黑暗中孵育30分钟。将40μL缓冲液中的不同浓度的苯基-哌嗪-三嗪添加至混合物中。在孵育15分钟之后,用200PRO微板读数仪(Tecan)测量荧光偏振(单位mP)。激发波长为485nm且在535nm处测量发射。根据常规方法(Wang,G.P.等,Abstr Pap Am Chem S 2004,228,U926-U926)计算Ki值。
7-3.实验结果
根据实施例6中的固相合成法,构建了苯基-哌嗪-三嗪化合物的化学库。为了模拟BH3螺旋肽的三个疏水残基(Leu、Val和Val),合成了在R2、R3和R6位置引入了多种疏水侧链的化合物。在36种库化合物中,选择化合物以在两种不同浓度(10μM和50μM)的化合物下对Mcl-1172-320(包含BH3结合槽的缺失构建物)与荧光标记的BH3肽之间的已知相互作用具有抑制活性。
如图16所示,PPT-31在10μM和50μM的两种浓度下对Mcl-1/BH3相互作用具有最高的抑制作用。如图17所示,PPT-31在每个R2、R3和R6处包含苯基(图17A),其预期位于能够与Mcl-1的疏水裂隙结合的BH3α-螺旋中Val220、Val216和Leu213的必需残基处(图17B)。通过使用相同的竞争性FP分析确定PPT-31的结合亲和力(Ki=7.3μM)(图17C)。
因为MCL-1的BH3结合口袋与Bcl-2家族蛋白(例如Bcl-xL)的BH3结合口袋相似,故本发明人测试了PPT-31的选择性。作为测试PPT-31对Bcl-xL的结合亲和力的结果,PPT-31不抑制BH3的Bcl-xL1-212(包含BH3结合口袋的缺失构建物)与BH3之间的相互作用(图17)。该结果表明PPT-31是MCL-1的选择性抑制剂。
制备实施例I.包含三嗪-哌嗪-三嗪支架的化合物的合成
如图3中所示,在5mL烧结注射器中用DMF(2mL)处理Rink-Amide MBHA树脂(100mg,93μmol)1小时。通过用DMF中的20%哌啶处理(2×10分钟)除去Fmoc基团。用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤树脂并将DMF(1mL)中的单取代的二氯三嗪(“3”)(5当量)与DIEA(5当量)在室温下孵育过夜以将单取代的二氯三嗪加载到树脂上。为了将由2-硝基苯磺酰基(Ns)保护的哌嗪衍生物(“5”)(10当量)与结合至树脂的三嗪衍生物(“4”)偶联,使产物在Et3N(5当量)的存在下在DMF中于60℃下反应过夜。除非另有说明,否则在每个反应步骤结束时用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤树脂。为了从偶联的化合物(“6”)中除去N-Ns保护基,使产物与2-巯基乙醇(20当量)和DBU(10当量)在DMF(10当量)中于室温下反应2小时。然后,为了引入2-乙基氢基-4,6-二氯-[1,3,5]三嗪(“7”)用于制备化合物(“8”),将化合物(e)(5当量)和DIEA(5当量)添加至NMP中的珠中并在60℃下过夜。用多种胺基团(R2’R2”NH)替代制备的化合物(“8”)的氯位置。最后,通过添加TFA(三氟乙酸)进行切割反应,以获得用三个官能团官能化的三嗪-哌嗪-三嗪化合物“9”。
用LC-MS确认由固相合成法制备的化合物的纯度和鉴定。最终产物的平均纯度为92%或更高,表明了固相合成法的效力。然后,用反相HPLC纯化化合物并用1H-NMR、13C-NMR和HRMS(高分辨率质谱仪)进行鉴定。
制备实施例1.化学式4化合物的制备
6-(4-(4-氨基-6-((苯并[d][1,3]二氧杂环戊烯-5-基甲基)氨基)-1,3,5-三嗪-2-基)-2-苄基哌嗪-1-基)-N2-苄基-N4-乙基-1,3,5-三嗪-2,4-二胺·TFA。
收率:来自100mg的树脂25.8mg(43%)。
1H-NMR(500MHz,甲醇-d4)δ1.26(m,3H)2.61-2.77(m,2H),3.05-3.15(m,2H),3.31-3.35(m,3H),4.31(m,1H),4.45-4.65(m,6H),5.04(m,1H),5.71(d,J=36.0Hz,1H),5.83(d,J=3.5Hz,1H),6.65-6.81(m,3H),6.98-7.08(m,5H),7.19(m,1H),7.25-7.27(m,4H);13C-NMR(126MHz,CDCl3)δ14.4,35.9,39.1,43.4,44.3,44.5,44.7,53.2,53.5,55.3,101.2,107.9,108.4,120.8,121.1,127.0,127.5,127.7,128.5,128.8,129.2,130.7,136.9,137.2,147.2,147.9,155.7,156.2,157.5,162.6;
HRMS(ESI)计算的C34H39N12O2[M+H]+:647.3319;实测:647.3319。
制备实施例2.化学式5化合物的制备
6-(4-(4-氨基-6-((环丙基甲基)(丙基)氨基)-1,3,5-三嗪-2-基)-2-苄基哌嗪-1-基)-N2-(苯并[d][1,3]二氧杂环戊烯-5-基甲基)-N4-乙基-1,3,5-三嗪-2,4-二胺·TFA。
收率:来自100mg树脂的18.8mg(31%)。
1H-NMR(500MHz,甲醇-d4)δ0.40(m,2H),0.62(m,2H),1.00(m,3H),1.12(br s,1H),1.25(t,J=7.5Hz,3H),1.72(m,2H),2.91-3.00(m,2H),3.33(m,2H),3.49-3.56(m,7H),4.46-4.61(m,4H),4.73(m,1H),5.19(m,1H),5.91(s,1H),5.94(s,1H).6.76-6.88(m,3H),7.13(d,J=6.5Hz,1H),7.20-7.25(m,4H);13C-NMR(126MHz,CDCl3)δ3.7,9.3,9.6,10.7,11.5,14.4,20.9,35.9,39.1,43.4,44.3,49.4,50.1,51.8,52.9,55.4,101.2,107.6,108.3,120.4,121.1,127.1,128.6,129.2,130.0,130.8,131.0,136.9,147.2,148.1,155.6,156.0,158.2,162.6;
HRMS(ESI)计算的C34H45N12O2[M+H]+:653.3788;实测:653.3787。
制备实施例3.化学式6所示化合物的制备
4-(2-((4-(4-(4-氨基-6-((4-苯氧基苯基)氨基)-1,3,5-三嗪-2-基)-2-苄基哌嗪-1-基)-6-(乙基氨基)-1,3,5-三嗪-2-基)氨基)乙基)苯酚·TFA。
收率:来自100mg树脂的27.0mg(41%)。
1H-NMR(500MHz,甲醇-d4)δ1.25(m,3H),2.82(m,2H),2.91(br s,2H),3.30(m,2H),3.49(m,3H),3.66(m,2H),4.57-4.68(m,3H),5.13(m,1H),6.73(m,2H),6.90(br s,1H),7.03-7.07(m,6H),7.17-7.24(m,5H)7.41(m,3H),7.58(br s,1H);13C-NMR(126MHz,DMSO-d6)δ14.6,14.8,34.2,35.7,36.0,42.5,43.2,44.1,53.9,115.6,118.7,119.5,122.9,123.7,126.7,128.7,129.2,129.6,130.0,130.1,130.5,138.2,152.5,155.1,155.4,156.3,157.6,162.1,163.8;
HRMS(ESI)计算的C39H43N12O2[M+H]+:711.3632;实测:711.3613。
制备实施例4.化学式7化合物的制备
6-(4-(4-氨基-6-(4-苯氧基苯基氨基)-1,3,5-三嗪-2-基)-2-苄基哌嗪-1-基)-N2-(苯并[d][1,3]二氧杂环戊烯-5-基甲基)-N4-乙基-1,3,5-三嗪-2,4-二胺·TFA。
1H-NMR(600MHz,CDCl3)δ1.28(m,3H)2.80-2.86(m,2H),3.14-3.22(m,2H),3.36-3.43(m,1H),3.48-3.53(m,2H),4.50-4.56(m,2H),4.63-4.77(m,3H),5.12(bs,1H),5.93-5.98(m,2H),6.76-6.89(m,4H),7.01-7.20(m,7H),7.26-7.29(m,1H),7.38-7.41(m,3H),7.58(dd,J=3.0,9.0Hz,1H),7.76-7.82(m,1H),8.05-8.16(m,1H),10.58(m,1H),14.59(bs,1H);13C-NMR(150MHz,CDCl3)δ14.3,35.9,36.2,39.1,39.2,43.6,44.2,44.5,53.4,101.2,107.6,108.4,118.9,120.4,121.0,123.2,123.7,127.1,128.6,129.0,129.3,129.8,131.3,136.7,147.2,148.1,154.2,154.7,155.8,156.2,157.0,157.6,162.5;
HRMS(ESI)计算的C39H41N12O3[M+H]+:725.3425;实测:725.3427。
制备实施例5.化学式8化合物的制备
6-(4-(4-氨基-6-(4-苯氧基苯基氨基)-1,3,5-三嗪-2-基)-2-苄基哌嗪-1-基)-N2-(3,4-二甲氧基苄基)-N4-乙基-1,3,5-三嗪-2,4-二胺·TFA(Q2)。
1H-NMR(600MHz,CDCl3)δ1.28(m,3H)2.81-2.86(m,2H),3.16-3.22(m,2H),3.36-3.43(m,1H),3.47-3.51(m,2H),3.80-3.91(m,5H),4.55-4.59(m,2H),4.65-4.69(m,2H),4.74-4.76(m,1H),5.12(bs,1H),6.81-6.89(m,4H),7.00-7.21(m,7H),7.26-7.29(m,1H),7.38-7.41(m,3H),7.58(dd,J=3.0,9.0Hz,1H),7.69-7.77(m,1H),8.23-8.30(m,1H),10.58(m,1H),14.31(bs,1H);13C-NMR(150MHz,CDCl3)δ14.3,35.9,36.3,39.2,43.6,44.3,44.5,53.4,55.9,110.6,110.1,111.3,118.9,119.1,119.4,119.9,123.2,123.6,127.1,128.6,129.0,129.3,129.9,131.3,136.7,148.7,149.3,154.2,154.7,155.7,156.0,157.0,157.5,162.5;
HRMS(ESI)计算的C40H40N12O3[M+H]+:741.3748;实测:741.3741。
制备实施例6.化合物9-nc的制备
6-(4-(4-氨基-6-(乙基氨基)-1,3,5-三嗪-2-基)哌嗪-1-基)-N2,N4-二乙基-1,3,5-三嗪-2,4-二胺·TFA。
收率:来自100mg树脂的18.0mg(50%)。
1H-NMR(500MHz,甲醇-d4)δ1.14(t,J=7.0Hz,9H),3.38(m,6H),3.89(br s,8H);13C-NMR(126MHz,DMSO-d6)δ14.6,35.7,43.4,155.8,157.4,162.1;
HRMS(ESI)计算的C16H29N12[M+H]+:389.2638;实测:389.2631。
制备实施例II.苯基-哌嗪-三嗪支架的化合物的合成
如图15所示,用DMF(2mL)处理Rink-Amide MBHA树脂(100mg,93μmol)1小时并用DMF中的20%哌啶处理(2×10分钟)以除去Fmoc基团。除非另有说明,否则在每个反应步骤结束时用DMF(3×)、MeOH(2×)、CH2Cl2(2×)和DMF(3×)彻底洗涤树脂。
将树脂添加至DMF(1.5mL)中的4-氟-3-硝基苯甲酸(70mg,378μmol)、HATU(142mg,378μmol)和DIEA(130μL,756μmol)中并将混合物在室温下孵育12小时。为了将硝基苯磺酰基保护的哌嗪(4当量)引入到树脂上,使产物与DIEA(10当量)在DMF中于95℃下反应12小时以产生树脂结合的哌嗪衍生物“4”。为了从衍生物“4”中除去硝基苯磺酰基,使产物与2-巯基乙醇(20当量)和DBU(10当量)在DMF中于室温下反应。为了将2-乙基氨基-4,6-二氯-[1,3,5]三嗪(10当量)偶联到树脂上,使产物与DIEA(5当量)在THF中于60℃下反应3小时以得到化合物“6”。为了用多种胺基团(R3NH2,5当量)取代化合物“6”的氯位置,使化合物与DIEA(10当量)在NMP中于80℃下反应过夜,以产生化合物“7”。将DMF中的SnCl2·2H2O(60当量)添加至树脂中并将混合物在室温下搅拌24小时。用混合溶液(CH(OMe)3∶DMF=9∶1)中的0.4M醛溶液(10当量)和甲醇中的20%(v/v)乙酸处理与树脂结合的胺(“8”)然后在50℃下反应18小时。然后,添加TMF中的5M NaCNBH3(50当量)并将混合物在50℃下搅拌6小时以产生烷基化产物。当使用脂肪醛时,添加苯并三唑(100当量)以防止二烷基化副产物的形成。最后,用TFA进行切割以产生用三个取代基官能化的苯基-哌嗪-三嗪衍生物。
用LC-MS分析由固相合成法制备的化合物的纯度和鉴定。结果,最终产物的平均纯度为90%或更高,证实了固相合成法的效力。用反相HPLC纯化化合物并用1H-NMR、13C-NMR和HRMS(高分辨质谱仪)进行鉴定。
制备实施例7.PPT-3化合物的制备
2-{4-(2-氨基乙基-4-氨基甲酰基苯基)-2-异丁基哌嗪}-4-氨基乙基-6-氨基苄基-[1,3,5]三嗪(PPT-3)。
1H NMR(300MHz,CDCl3)δ0.92(t,J=6.9Hz,3H),0.97(dd,J=6.3,9.3Hz,3H),1.20(q,J=7.2Hz,3H),1.33(t,J=6.6Hz,3H),1.55-1.62(m,1H),1.76-1.91(m,2H),2.60-2.84(m,2H),3.05-3.32(m,5H),3.42-3.48(m,2H),4.61(t,J=5.7Hz,2H),4.84(t,,J=14.7Hz,1H),4.98-5.03(m,1H),6.93(d,J=8.1Hz,1H),7.07-7.13(m,2H),7.24-7.37(m,4H),7.60-7.62(m,1H),7.97-8.00(m,1H);13C NMR(75MHz,CDCl3)δ16.1,16.2,16.6,24.4,24.5,24.9,26.7,37.7,40.4,40.7,41.8,46.0,46.1,46.3,52.0,52.7,55.9,111,4,117.9,121.1,129.0,129.5,129.8,132.1,139.2,143.7,145.0,157.4,157.9,164.1,173.0.
HRMS(FAB)计算的C29H42N9O[M+H]+:532.3512;实测:532.3509。
制备实施例8.PPT-5化合物的制备
2-{4-(2-氨基乙基-4-氨基甲酰基苯基)-2-苄基哌嗪}-4-氨基乙基-6-氨基异丙基-[1,3,5]三嗪(PPT-5)。
1H NMR(300MHz,CDCl3)δ1.20-1.30(m,6H),1.36(t,J=6.9Hz,6H),2.69-2.78(m,1H),2.83(dd,J=3.9,12.0Hz,1H),3.11-3.54(m,9H),4.07-4.20(m,1H),4.79-4.86(m,1H),5.17(brs,1H),6.94(d,J=8.1Hz,1H),7.05(dd,J=1.8,8.1Hz,1H),7.16-7.43(m,6H);13C NMR(75MHz,CDCl3)δ16.1,16.3,16.8,24.0,24.2,37.6,40.2,42.2,45.1,53.1,54.6,55.2,111.4,117.7,121.2,130.4,130.9,131.0,131.6,139.7,143.5,145.1,156.6,157.3,164.3,164.6,172.7.
HRMS(FAB)计算的C28H39N9O[M+H]+:518.3356;实测:518.3355。
制备实施例9.PPT-15的化合物的制备
2-{4-(2-氨基异丁基-4-氨基甲酰基苯基)-2-异丁基哌嗪}-4-氨基乙基-6-氨基苄基-[1,3,5]三嗪(PPT-15)。
1H NMR(300MHz,CDCl3)δ0.89(dd,J=3,6.6Hz,3H),0.98(t,J=6.9Hz,3H),1.03(dd,J=3,6.6Hz,6H),1.20-1.26(m,3H),1.50-1.57(m,1H),1.67-1.74(m,1H),1.92-2.04(m,2H),2.67-2.83(m,2H),2.98-3.12(m,4H),3.31-3.49(m,3H),4.58-4.62(m,2H),4.78(t,J=15.0Hz,1H),5.02-5.05(m,1H),6.93(d,J=8.1Hz,2H),7.06-7.11(m,2H),7.28-7.33(m,2H),7.60-7.63(m,1H),8.01-8.03(m,1H);13C NMR(75MHz,CDCl3)δ12.3,12.4,18.7,20.8,21.0,22.8,26.2,33.9,37.0,38.0,42.4,42.6,48.1,48.2,49.2,49.8,52.2,107.6,113.9,117.4,125.3,125.7,126.8,127.1,135.2,135.4,140.1,141.5,153.5,153.8,160.1,169.8.
HRMS(FAB)计算的C31H46N9O[M+H]+:560.3825;实测:560.3827。
制备实施例10.PPT-19的化合物的制备
2-{4-(2-氨基异丁基-4-氨基甲酰基苯基)-2-苄基哌嗪}-4-氨基乙基-6-氨基苄基-[1,3,5]三嗪(PPT-19)。
1H NMR(300MHz,CDCl3)δ1.06-1.08(m,6H),1.20-1.26(m,3H),1.96-2.05(m,1H),2.57-2.68(m,2H),2.84-2.94(m,1H),3.06-3.23(m,5H),3.35-3.49(m,3H),4.50-4.65(m,2H),4.78(t,J=15.0Hz,1H),5.10-5.16(m,1H),6.90(dd,J=1.8,8.1Hz,2H),6.98-7.04(m,2H),7.11-7.34(m,7H),7.57-7.65(m,1H),7.96-8.03(m,1H);13C NMR(75MHz,CDCl3)δ13.9,14.0,20.2,27.8,35.4,35.6,40.1,44.0,51.1,51.9,109.0,115.0,118.8,126.4,127.1,128.2,128.3,128.7,129.8,137.3,140.8,143.1,155.6,162.1,169.9.
HRMS(FAB)计算的C34H43N9O[M+H]+:594.3669;实测:594.3665。
制备实施例11.PPT-23的化合物的制备
2-{4-(2-氨基异丁基-4-氨基甲酰基苯基)-2-甲基哌嗪}-4-氨基乙基-6-氨基苄基-[1,3,5]三嗪(PPT-23)。
1H NMR(300MHz,CDCl3)δ1.05(q,J=3.3Hz,6H),1.26(t,J=7.2Hz,3H),1.38(dd,J=6.9,25.5Hz,3H),1.95-2.07(m,1H),2.74-2.85(m,2H),2.97-3.18(m,4H),3.32-3.50(m,3H),4.52-4.63(m,3H),4.88-5.02(m,1H),6.95(d,J=8.1Hz,1H),6.98-7.09(m,2H),7.28(d,J=2.7Hz,3H),7.67-7.74(m,1H),8.12-8.21(m,1H);13C NMR(75MHz,CDCl3)δ16.1,17.3,22.5,30.1,37.7,41.6,46.4,49.4,52.8,53.5,57.3,111.2,117.3,120.8,129.4,130.5,131.8,143.3,145.2,172.1.
HRMS(FAB)计算的C28H40N9O[M+H]+:518.3356;实测:518.3353。
制备实施例12.PPT-28化合物的制备
2-{4-(2-氨基苄基-4-氨基甲酰基苯基)-2-异丁基哌嗪}-4-氨基乙基-6-氨基-2-萘基甲基-[1,3,5]三嗪(PPT-28)。
1H NMR(300MHz,CDCl3)δ0.61(dd,J=6.0,14.7Hz,3H),0.78(dd,J=6.0,14.7Hz,3H),1.18-1.26(m,3H),1.30-1.44(m,2H),1.72-1.81(m,1H),2.60-2.86(m,2H),3.01-3.49(m,5H),4.27-4.43(m,2H),4.68-5.07(m,4H),6.93(dd,J=4.2,7.8Hz,1H),7.10-7.14(m,2H),7.31-8.04(m,12H);13C NMR(75MHz,CDCl3)δ13.8,22.1,24.3,35.5,38.2,38.3,39.5,41.3,48.1,49.7,49.8,50.5,53.6,53.9,109.2,116.1,117.2,118.9,122.4,122.6,124.9,125.6,126.1,127.4,128.1,128.5,128.7,130.6,130.8,131.8,132.1,133.5,138.2,141.7,142.6,155.1,155.3,155.5,161.6,161.7,162.1,171.0.
HRMS(FAB)计算的C38H46N9O[M+H]+:644.3825;实测:644.3827。
制备实施例13.PPT-31化合物的制备
2-{4-(2-氨基苄基-4-氨基甲酰基苯基)-2-苄基哌嗪}-4-氨基乙基-6-氨基苄基-[1,3,5]三嗪(PPT-31)
1H NMR(300MHz,CDCl3)δ1.18-1.25(m,3H),2.63-3.14(m,6H),3.22-3.31(m,1H),3.36-3.45(m,2H),4.45(d,J=2.7Hz,2H),4.53-4.64(m,2H),4.76(t,J=13.8Hz,1H),5.06-5.12(m,1H),6.91-7.37(m,16H),7.57-7.68(m,1H),7.96-8.03(m,1H);13C NMR(75MHz,CDCl3)δ14.4,35.8,40.4,44.1,44.2,44.4,48.1,51.2,51.3,52.3,52.4,53.2,53.3,109.7,116.3,119.4,126.7,127.3,127.6,128.6,128.9,129.0,130.0,137.6,138.9,141.5,143.0,155.5,156.0,162.4,170.4.
HRMS(FAB)计算的C37H42N9O[M+H]+:628.3512;实测:628.3510。
制备实施例14.PPT-36化合物的制备
2-{4-(2-氨基苄基-4-氨基甲酰基苯基)-2-甲基哌嗪}-4-氨基乙基-6-氨基-2-萘基甲基-[1,3,5]三嗪(PPT-36)。
1H NMR(300MHz,CDCl3)δ1.08-1.24(m,6H),2.65-2.84(m,2H),3.03-3.29(m,3H),3.40(quin,J=6.8Hz,2H),4.28(q,J=13.5Hz,2H),4.65(t,J=13.5Hz,1H),4.87-5.10(m,3H),6.93(d,J=8.7Hz,1H),7.09-7.12(m,2H),7.30-7.52(m,8H),7.76-7.87(m,1H),8.01-8.17(m,1H);13C NMR(75MHz,CDCl3)δ16.0,17.1,37.8,44.1,49.4,50.3,52.6,57.1,111.4,118.5,120.9,124.8,127.2,127.8,128.0,128.3,129.5,129.6,130.4,130.7,130.8,133.0,134.4,135.7,140.4,140.8,144.1,144.6,157.6,163.5,163.6,164.0,173.5.
HRMS(FAB)计算的C35H40N9O[M+H]+:602.3356;实测值:602.3358。
- 还没有人留言评论。精彩留言会获得点赞!