一种吡啶衍生物的多晶型及其制备方法、用途与流程
本发明涉及药物化学合成领域,具体涉及化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物的多晶型及其制备方法及其作为治疗药物,特别是作为在制备预防或治疗血栓形成或栓塞的药物中的用途。
背景技术:
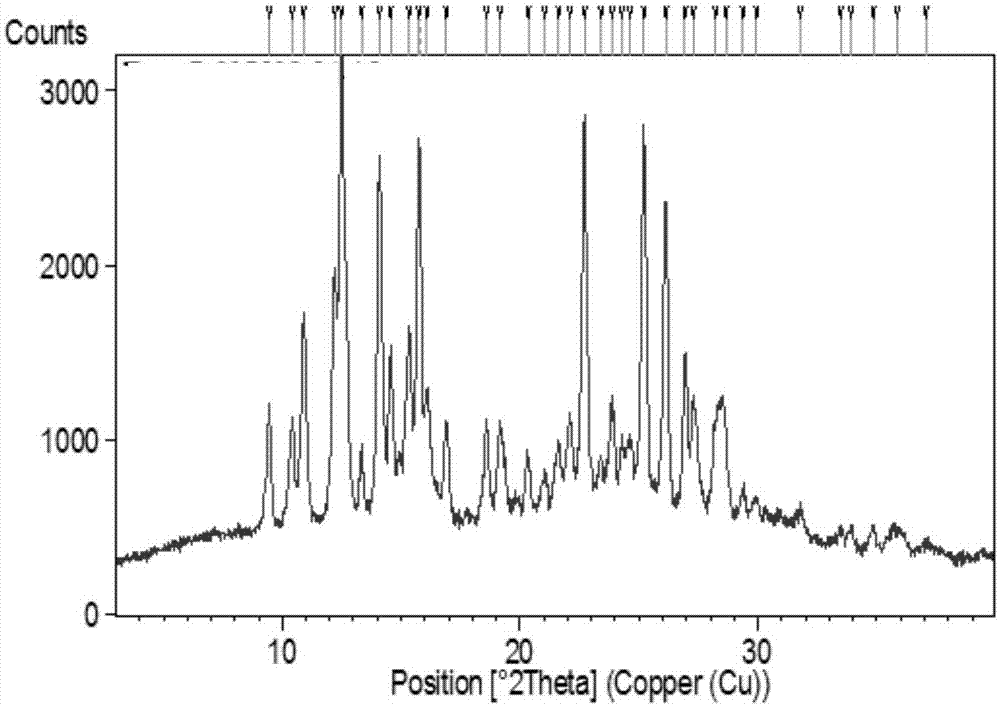
:血栓性疾病是严重危害人类健康的疾病,根据血栓形成部位、条件与性质,主要分为动脉血栓与静脉血栓。动脉血栓形成是从动脉血管壁动脉粥样硬化病变与血小板激活开始,其导致的严重临床反应主要为急性心肌梗死、脑卒中;静脉血栓由静脉血管中多种原因诱发形成,可导致静脉血栓栓塞(venousthromboembolism,VTE),其主要临床表现为深静脉血栓形成(deepvenousthrombosis,DVT)和肺栓塞(pulmonaryembolism,PE)。VTE是继急性冠脉综合征与脑卒中之后的第三大心血管疾病。在医院所有死亡病例中,VTE约占10%,欧盟6国中每年症状性VTE发生总数100万,死亡病例数超过艾滋病、乳腺癌、前列腺癌及交通事故造成死亡的总和。美国死亡病例则超过29.6万/年,而致死性PE在死亡前仅有不到50%得到确诊。国际上的相关指南已将预防VTE列为降低住院患者死亡率最重要的策略之一。大规模临床试验证据显示,抗凝治疗可阻止血栓的蔓延和复发,并进一步降低卒中、PE等的发生率和死亡率。因此,抗凝治疗已成为目前临床预防和治疗血栓栓塞性疾病的核心和基础,而抗凝药物的研发也始终是新药研发的热点,尤其是以Xa因子抑制剂为靶点的药物研发。本发明在不断地研究过程中,通过试验摸索,意外的获得了新的1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物的多晶型。众所周知,对于药物的多晶型而言,不同的晶型可以具有不同的化学和物理特性,包括化学稳定性、溶解度、光学和机械性质等,这些性质可以直接影响原料药和制剂的处理和生产工艺过程,并且会影响到制剂的稳定性、溶解度和生物利用度,因此,多晶型的研究对于药物制剂的质量、安全性和有效性具有重要意义。对于Xa因子抑制剂而言,本领域存在着这样的更适合于工业化生产、理化性质优异的1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物的多晶型。技术实现要素:本发明公开了一种如下结构式I所示的1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物的多晶型,以及各晶型制备方法,及其各晶型在制备对Xa因子抑制剂相关疾病进行治疗的药物中的用途。更具体而言,所述用途为在制备预防或治疗血栓或栓塞药物中的用途。本发明公开的式I结构化合物的多晶型具体包括晶型B、M1、O、P、Q、T、U、V和X。本发明提供了式I化合物的晶型B,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型B的谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型B具有基本如附图2所示的X射线粉末衍射图谱。本发明提供了式I化合物的晶型M1,使用Cu-ka射线进行X射线粉末测定,图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型M1的谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型M1具有基本如附图3所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型O,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型O的谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型O具有基本如附图4所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型P,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型P谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型P具有基本如附图5所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型Q,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型Q的谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型Q具有基本如附图6所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型T,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型T谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型T具有基本如附图7所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型U,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型U谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型U具有基本如附图8所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型V,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型V谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型V具有基本如附图9所示的X射线粉末衍射图谱。本发明又一方面提供了式I化合物的晶型X,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。进一步的,晶型X谱图具有下表所示的衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。更进一步的,晶型X具有基本如附图10所示的X射线粉末衍射图谱。本发明还公开的式I结构化合物的多晶型具体包括晶型C、D、F、H、I、J、L、M、R和S。本发明又一方面提供了式I化合物的晶型C,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度。2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型D,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型F,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型H,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型I,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度。2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型J,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型L,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型M,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度。2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型R,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明又一方面提供了式I化合物的晶型S,使用Cu-ka射线进行X射线粉末测定,其图谱具有下表所示衍射角、晶面间距和相对强度:2θ衍射角的误差为±0.2。本发明还公开了式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的制备方法:在室温(10~25℃)条件下,起始原料2-甲基-4-硝基苯胺与5-氯代戊酰氯反应生成化合物a,化合物a在碱性条件下发生分子内关环生成化合物b,化合物b在五氯化磷的作用下生成化合物c,生成的化合物c进一步在吗啉的作用下生成化合物d,化合物d中的硝基还原后生成化合物e,化合物e再一次与4-氯代丁酰氯反应生成化合物f,化合物f再一次发生分子内关环生成化合物g,化合物g与[(4-甲氧基苯基)肼基]氯乙酸乙酯生成化合物h,化合物h胺解后得到式I化合物。本发明式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的晶型B、M1、O、P、Q、T、U、V和X可以通过如下方法制备:(1)将式I结构所示的化合物加入到样品瓶中,加入正溶剂,在室温或回流条件下使其完全溶解,直接或滴加反溶剂后冷却至室温结晶;(2)抽滤,室温或加热条件下常压或真空干燥,得到不同的晶型;以及任选的,将该晶型加在反溶剂中形成悬浊液后搅拌、抽滤、干燥获得不同晶型。其中,正溶剂是指本发明的结构式I化合物在该溶剂中溶解性较好,正溶剂具体选自甲醇、乙醇、四氢呋喃、丙酮、N,N-二甲基甲酰胺、二氧六环、二氯甲烷和和乙酸乙酯中一种或多种;优选甲醇、乙醇、二氯甲烷、N,N-二甲基甲酰胺和和乙酸乙酯中一种或多种;反溶剂是指本发明的结构式I化合物在该溶剂中溶解性较差,反溶剂具体选自水、甲基叔丁基醚、正庚烷、正己烷和环己烷中一种或多种;优选水、甲基叔丁基醚和正庚烷中一种或多种。进一步,本发明式I化合物的晶型B、M1、O、P、Q、T、U、V和X可以通过如下方法制备:(1)将式I结构所示的化合物加入到样品瓶中,加入正溶剂,在室温或回流条件下使其完全溶解,直接或滴加反溶剂后冷却至室温结晶;(2)抽滤,室温或加热条件下常压或真空干燥,得到不同的晶型。或者,进一步,本发明式I化合物的晶型B、M1、O、P、Q、T、U、V和X可以通过如下方法制备:(1)将式I结构所示的化合物加入到样品瓶中,加入正溶剂,在室温或回流条件下使其完全溶解,直接或滴加反溶剂后冷却至室温结晶;(2)抽滤,室温或加热条件下常压或真空干燥,得到不同的晶型,将该晶型加在反溶剂中形成悬浊液后搅拌、抽滤、干燥获得不同晶型。本发明通过溶解度试验发现,式I化合物的晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V或晶型X都具有很好的溶解性;通过5天和10天的化学稳定性考察试验发现,式I化合物的晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V和晶型X均具有优异的化学稳定性。进一步的,通过10天的物理稳定性考察试验发现,本发明晶型具有良好的物理稳定性。再进一步,本发明考察了式I化合物的无定形、晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V和晶型X在常温常湿条件下的吸潮性,结果显示本发明晶型B、晶型X无吸湿性,晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型V略有吸湿性,晶型U有吸湿性。通过对正常小鼠APTT的作用试验发现,本发明制备的无定型及各种晶型具有显著延长小鼠APTT值的作用,均显著优于阳性药阿哌沙班;而与无定型相比,本发明B晶型的APTT值极显著增加(P<0.01),晶型M1、O、Q、X晶型的APTT值显著增加(P<0.05);说明晶型B、晶型M1、晶型O、晶型Q、晶型X的效果相对更好。即本发明制备的式I化合物的晶型B、M1、O、Q、X可用于制备抗凝、预防或治疗血栓或栓塞药物中的用途,特别是在制备抗凝药物中的用途或制备Xa因子抑制剂药物中的用途。附图说明图1式I化合物无定型的XRPD图谱图2式I化合物晶型B的XRPD图谱图3式I化合物晶型M1的XRPD图谱图4式I化合物晶型O的XRPD图谱图5式I化合物晶型P的XRPD图谱图6式I化合物晶型Q的XRPD图谱图7式I化合物晶型T的XRPD图谱图8式I化合物晶型U的XRPD图谱图9式I化合物晶型V的XRPD图谱图10式I化合物晶型X的XRPD图谱具体实施方式以下将结合实施例对本发明作进一步的详细描述,本发明的实施例仅用于说明本发明的技术方案,并非限制本发明的实质和范围。化合物的结构是通过核磁共振(1HNMR)来测定的。核磁共振(1HNMR)位移(δ)以百万分之一(ppm)为单位给出;核磁共振(1HNMR)的测定是用BrukerAVANCE-400核磁仪进行,测定溶剂为六氘代二甲基亚砜(CDCl3),内标为四甲基硅烷(TMS)。质谱(MS)的测定用FINNIGANLCQAd(ESI)质谱仪(生产商:Therm,型号:FinniganLCQadvantageMAX)进行。HPLC谱图的测定采用安捷伦Agilent1260DAD型液相色谱仪进行。在本发明的术语“氮气氛”是指例如将反应瓶连接一个1L容积的氮气气球。薄层硅胶使用烟台黄海HSGF254或青岛GF254硅胶板。X射线粉末衍射(XRPD)的测定是采用PanalyticalEmpyreanX射线粉末衍射分析仪进行,具体参数见下表:热重分析(TGA)和差示扫描量热仪(DSC)测定数据分别在TAQ500热重分析和TAQ200差示扫描量热仪上采集,仪器参数列于下表:本发明中的术语“室温”是指温度处于10℃至25℃之间。实施例1化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的制备制备方案如下所示:第一步:a的制备将2-甲基-4-硝基苯胺(3g,19.7mmol)溶于二氯甲烷(60ml)中,加入N,N-二异丙基乙胺(6.4g,49.5mmol),冰浴降温到5℃以下,滴加5-氯乙酰氯(3.7g,23.9mmol)以得到反应混合物,使所述反应混合物在室温下反应过夜。薄层色谱跟踪反应进程,反应完全后,向反应液中倒入水、分液、水洗3次、无水硫酸镁干燥,过滤,减压蒸馏除去溶剂后得化合物a(4.2g,黄色固体),收率:78.8%。MSm/z(ES):271.1[M+1]第二步:b的制备将a(4.2g,15.5mmol)溶于四氢呋喃(80ml)中,冰浴下分批加入氢化钠(0.75g,31.3mmol)以得到反应混合物,使所述反应混合物在室温下反应过夜。薄层色谱跟踪反应进程,反应完全后,冰浴下向反应混合物中加入冰水,淬灭氢化钠,减压蒸馏除去四氢呋喃后,乙酸乙酯萃取2次、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后残留物经柱层析纯化得b(3.27g,黄色固体),收率:90.1%。MSm/z(ES):235.1[M+1]第三步:c的制备将b(3.27g,14.0mmol)溶于二氯甲烷(100ml)中,冰浴下分批加入五氯化磷(8.7g,41.8mmol)以得到反应混合物,使所述反应混合物在40℃下回流反应。待反应液基本不冒气泡时,薄层色谱跟踪反应进程,反应完全后,冰浴下向反应混合物中加入冰水淬灭五氯化磷、分液、水洗3次、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后得c(4g,黄色固体),收率:94.6%。MSm/z(ES):303.0,305.0[M+1]第四步:d的制备将c(4g,13.2mmol)溶于吗啉(40ml)中以得到反应混合物,使所述反应混合物在120℃下回流反应2小时,薄层色谱跟踪反应进程,反应完全后,加入乙酸乙酯溶解、水洗3次、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后得d(3.98g,黑色固体),收率:95.0%。MSm/z(ES):318.1[M+1]第五步:e的制备将d(3.98g,12.5mmol)溶于乙醇(50ml)中,加入九水硫化钠(9g,37.5mmol),再加入水(20ml)以得到反应混合物,使所述反应混合物在50℃下回流反应过夜。薄层色谱跟踪反应进程,反应完全后,减压浓缩除去乙醇、乙酸乙酯萃取三次、合并有机相、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后得e(3.2g,黄色固体),收率:88.9%。MSm/z(ES):288.2[M+1]第六步:f的制备将e(3.2g,11.1mmol)溶于二氯甲烷(50ml)中,加入N,N-二异丙基乙胺(3.6g,27.9mmol),冰浴下,滴加4-氯丁酰氯(2.4g,17.0mmol)以得到反应混合物,使所述反应混合物在室温下反应过夜。薄层色谱跟踪反应进程,反应完全后,向反应液中加入水、分液、水洗3次、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后得f(3.5g,黄色固体),收率:80.3%。MSm/z(ES):392.2[M+1]第七步:g的制备将f(3.5g,8.9mmol)溶于四氢呋喃(50ml)中,冰浴下分批加入氢化钠(0.6g,25mmol)以得到反应混合物,使所述反应混合物在室温下反应过夜。薄层色谱跟踪反应进程,反应完全后,向反应液中加入冰水淬灭氢化钠,减压蒸馏除去四氢呋喃后,加入二氯甲烷萃取2次、合并有机相、无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后,残留物经柱层析纯化得g(2.59g,黄色固体),收率:81.7%。MSm/z(ES):356.2[M+1]第八步:h的制备将g(280mg,0.79mmol)溶于甲苯(10ml)中,室温下加入[(4-甲氧基苯基)肼基]氯乙酸乙酯(214mg,0.83mmol)、三乙胺(252mg,2.5mmol)以得到反应混合物,使所述反应混合物在120℃下回流反应过夜。薄层色谱跟踪反应进程,反应完全后,减压蒸馏除去甲苯,残留物加入二氯甲烷(20ml)溶解后,室温下加入三氟乙酸(2ml)以得到反应混合物,使所述反应混合物在室温下反应过夜。薄层色谱跟踪反应进程,反应完全后,经柱层析纯化得h(300mg,黄色固体),收率:77.9%。MSm/z(ES):489.2[M+1]第九步:结构式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的制备将h(300mg,0.61mmol)溶于甲醇(4ml)中,加入氨水(2ml)以得到反应混合物,使所述反应混合物在70℃下回流反应过夜。薄层色谱跟踪反应进程,反应完全后,经柱层析纯化得结构式I化合物(208mg,浅黄色固体),收率:73.8%。MSm/z(ES):460.2[M+1]1HNMR(400MHz,CDCl3)δ7.51–7.46(m,4H),7.17(d,J=8.5Hz,1H),6.93(d,J=8.8Hz,2H),6.87(brs,1H),5.54(brs,1H),4.09–4.02(m,1H),3.84–3.79(m,6H),3.45–3.32(m,2H),2.60(t,J=8.0Hz,2H),2.26(s,3H),2.19–2.13(m,2H).将柱层析得到的样品用X-射线粉末衍射(XRPD)图像进行固态分析,结果发现该样品在XRPD中无明显特征峰,故判定该样品为无定型,见附图1。通过薄层色谱分离得到的产品进行了XRPD法分析,发现样品仍然是无定型。实施例2式I化合物的晶型B的制备将100mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入3ml甲醇,加热使其完全溶解后,冷却至室温,抽滤,收集析出的晶体,150℃真空干燥,得到82mg浅黄色固体,收率82%。该结晶样品的X-射线粉末衍射图谱见附图2,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约202℃有特征吸收峰,TGA显示该结晶样品加热至150℃时失重为0.25%。根据以上结果得知,该晶型为无水晶型,定义为晶型B。实施例3式I化合物的晶型M1的制备将30mg按照实施例2制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的晶型B加入样品瓶中,加入0.5ml丙酮,形成的悬浊液室温搅拌过夜,离心分离后得到的样品室温干燥,得到26mg浅黄色固体,收率86.7%。该结晶样品的X-射线粉末衍射图谱见附图3,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约85℃、106℃、144℃、149℃、179℃和208℃有多个特征吸热/放热峰,TGA显示该结晶样品加热至100℃时失重为7.40%。该结晶样品的1HNMR测试结果表明样品中含丙酮质量分数0.2%,结合加热试验所得TGA数据,可推测丙酮为吸附溶剂。对该结晶样品用KF法进行水分测定得出其含水量为7.2%。根据以上结果得知,该晶型为二水合物,定义为晶型M1。实施例4式I化合物的晶型O的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入1ml二氯甲烷将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至180℃后泠却至室温,得到25mg浅黄色固体,收率83.3%。该结晶样品的X-射线粉末衍射图谱见附图4,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约199℃和208℃有重叠的吸热峰,TGA显示该结晶样品加热至180℃时失重为0.82%。根据以上结果得知,该晶型为无水晶型,定义为晶型O。实施例5式I化合物的晶型P的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入1ml二氯甲烷将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至205℃后泠却至室温,得到24mg浅黄色固体,收率80.0%。该结晶样品的X-射线粉末衍射图谱见附图5,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约211℃有特征吸收峰,TGA显示该结晶样品加热至200℃时失重为0.24%。根据以上结果得知,该晶型为无水晶型,定义为晶型P。实施例6式I化合物的晶型Q的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入1ml二氯甲烷将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至140℃后泠却至室温,得到26mg浅黄色固体,收率86.7%。该结晶样品的X-射线粉末衍射图谱见附图6,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约156℃、168℃、189℃、200℃和208℃有多个吸热/放热峰,TGA显示该结晶样品加热至150℃时失重为1.1%。根据以上结果得知,该晶型为无水晶型,定义为晶型Q。实施例7式I化合物的晶型T的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物加入样品瓶中,加入0.5ml四氢呋喃,形成的悬浊液在50℃搅拌过夜,离心分离后室温真空干燥。将所得的固体溶于1.5ml纯水中,形成的悬浊液在50℃搅拌过夜,离心分离后室温真空干燥后,得到22mg浅黄色固体,收率73.3%。该结晶样品的X-射线粉末衍射图谱见附图7,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约83℃、107℃、148℃和208℃有多个特征吸热/放热峰,TGA显示该结晶样品加热至120℃时失重为7.7%。该结晶样品的1HNMR测试结果表明样品中含四氢呋喃质量分数0.3%。对该结晶样品用KF法进行水分测定得出其含水量为7.3%。根据以上结果得知,该晶型为二水合物,定义为晶型T。实施例8式I化合物的晶型U的制备将15mg按照实施例7制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的晶型T在氮气氛围中吹扫40min后,得到13.5mg浅黄色固体,收率90.0%。该结晶样品的X-射线粉末衍射图谱见附图8,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品重新暴露在空气中后,可迅速吸水转变为晶型T。根据以上结果得知,该晶型为无水晶型,定义为晶型U。实施例9式I化合物的晶型V的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物加入样品瓶中,加入0.9ml甲醇将其完全溶解,室温下缓慢挥发使其析出固体。离心分离后得到固体在氮气保护下加热到120℃后泠却至室温,得到18mg浅黄色固体,收率60.0%。该结晶样品的X-射线粉末衍射图谱见附图9,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约168℃、175℃和208℃有多个特征吸热/放热峰,TGA显示该结晶样品加热至15℃时失重为1.9%。该结晶样品的1HNMR测试结果表明样品中含甲醇质量分数0.2%。对该结晶样品用KF法进行水分测定得出其含水量为2.1%。根据以上结果得知,该晶型为水合物晶型,定义为晶型V。实施例10式I化合物的晶型X的制备将200mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入1.6mlN,N-二甲基甲酰胺,加热到80℃使其完全溶解后,滴加1.2ml水,自然泠却至室温,抽滤,收集析出的晶体,70℃干燥,得到166mg浅黄色固体,收率83.0%。该结晶样品的X-射线粉末衍射图谱见附图10,其图谱具有下表所示衍射角、晶面间距和相对强度的特征峰:该结晶样品的DSC在约214℃有特征吸收峰,TGA显示该结晶样品加热至150℃时失重为0.32%。根据以上结果得知,该晶型为无水晶型,定义为晶型X。实施例11式I化合物的晶型B的制备将100mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入10ml乙酸乙酯,加热使其完全溶解后,冷却至室温,抽滤,收集析出的晶体,150℃真空干燥,得到86mg浅黄色固体,收率86%。粉末衍射图谱与实施例2保持一致。实施例12式I化合物的晶型M1的制备将50mg按照实施例2制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的晶型B加入样品瓶中,加入0.45ml四氢呋喃,形成的悬浊液室温搅拌过夜,离心分离后得到的样品室温干燥,得到42mg浅黄色固体,收率84%。粉末衍射图谱与实施例3保持一致。实施例13式I化合物的晶型O的制备将40mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入0.5ml四氢呋喃和0.6ml乙酸乙酯将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至180℃后泠却至室温,得到34mg浅黄色固体,收率85%。粉末衍射图谱与实施例4保持一致。实施例14式I化合物的晶型P的制备将40mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入0.5ml四氢呋喃和0.6ml乙酸乙酯将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至205℃后泠却至室温,得到28mg浅黄色固体,收率70%。粉末衍射图谱与实施例5保持一致。实施例15式I化合物的晶型Q的制备将40mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入0.5ml四氢呋喃和0.6ml乙酸乙酯将其溶解,室温下缓慢挥发使其析出固体,抽滤,将所得的固体在氮气保护下加热至140℃后泠却至室温,得到30mg浅黄色固体,收率75%。粉末衍射图谱与实施例6保持一致。实施例16式I化合物的晶型T的制备将30mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物加入样品瓶中,加入0.6ml丙酮,形成的悬浊液在50℃搅拌过夜,离心分离后室温真空干燥。将所得的固体溶于1.5ml水,形成的悬浊液在50℃搅拌过夜,离心分离后室温真空干燥后,得到24mg浅黄色固体,收率80%。粉末衍射图谱与实施例7保持一致。实施例17式I化合物的晶型U的制备将20mg按照实施例16制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺的晶型T在氮气氛围中吹扫40min后,得到18.5mg浅黄色固体,收率92.5%。粉末衍射图谱与实施例8保持一致。实施例18式I化合物的晶型V的制备将20mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺化合物加入样品瓶中,加入0.7ml二氧六环将其完全溶解,室温下缓慢挥发使其析出固体。离心分离后得到固体在氮气保护下加热到120℃后泠却至室温,得到14mg浅黄色固体,收率70.0%。粉末衍射图谱与实施例9保持一致。实施例19式I化合物的晶型X的制备将100mg按照实施例1制备的式I化合物1-(4-甲氧基苯基)-7-氧代-6-[2-甲基-4-(2-氧代四氢吡咯-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺加入样品瓶中,加入0.9mlN,N-二甲基甲酰胺和0.5ml水,加热到80℃使其完全溶解后,自然泠却至室温,抽滤,收集析出的晶体,70℃干燥,得到73mg浅黄色固体,收率73.0%。粉末衍射图谱与实施例10保持一致。试验例1溶解性考察试验为了考察本发明实施例1制备的无定形与实施例2~10制备的晶型B、M1、O、P、Q、T、U、V和X的溶解性,本发明分别在25℃、37℃条件下在pH=1.0(0.1N)的盐酸和pH=4.0的乙酸-乙酸钠缓冲溶液中,测定实施例1制备的无定形,实施例2~10制备的晶型B、M1、O、P、Q、T、U、V和X的平衡溶解度(饱和溶液),结果如下表1所示:表1溶解性试验溶解性试验结果表明,25℃、37℃条件下在pH=1.0(0.1N)的盐酸和pH=4.0的乙酸-乙酸钠缓冲溶液中,式I化合物的无定型、晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V和晶型X都具有很好的平衡溶解性。试验例2化学稳定性考察试验将实施例1制备无定型和实施例2~10制备的晶型B、M1、O、P、Q、T、U、V和X样品分别放入洁净的培养皿中敞口平摊放置,考察在高温(60℃)、高湿(25℃、RH90%±5%)、强光(4500Lx±500Lx)条件下样品的稳定性,考察取样时间为5天和10天,放置10天。分别在5天和10天取样测定,HPLC纯度检测结果见下表2:表2稳定性试验(纯度%)稳定性考察结果表明,式I结构所示的无定型、晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V和晶型X在敞口放置的条件下,经高温(60℃)、高湿(25℃、RH90%±5%)、强光(4500Lx±500Lx)等条件下的稳定性比较发现,本发明晶型B、晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型U、晶型V和晶型X在高湿、高温、光照条件纯度无明显变化,即本发明各种晶型的化学稳定性均较好;而无定型产物在高湿条件下的纯度无明显变化,但高温条件下和光照条件下纯度均明显下降。说明在光照、高温、高湿条件下,本发明实施例2~10制备的晶型B、M1、O、P、Q、T、U、V和X的稳定性明显优于无定型。试验例3物理稳定性考察试验实施例2制备的样品晶型B(作为晶型B0天参考数据,批号20140408),实施例5制备的样品晶型P(作为晶型P0天参考数据,批号20140415),实施例6制备的样品晶型Q(作为晶型Q0天参考数据,批号20140418),实施例7制备的样品晶型T(作为晶型T0天参考数据,批号20140421),实施例10制备的样品晶型X(作为晶型X0天参考数据,批号20140427),分别放入洁净的培养皿中敞口平摊放置,考察在高温(60℃)、高湿(25℃、RH90%±5%)、强光(4500Lx±500Lx)条件下,各晶型样品的物理稳定性,考察取样时间为10天。在第10天取样,用X-射线粉末衍射(XRPD)图进行固态分析,粉末衍射数据结果如下表3~7所示。结果表明,在敞口放置的条件下,经高温、高湿、强光等条件下,10天后,晶型B、P、Q、T、X的X-射线粉末衍射角度值(以2θ衍射角表示)与样品晶型B、P、Q、T、X0天的参考数据均保持一致,即说明本发明晶型的物理稳定性好。表3晶型B在高温、高湿、光照条件下的粉末衍射角度数据(2θ衍射角)表4晶型P在高温、高湿、光照条件下的粉末衍射角度数据(2θ衍射角)PeakNo.晶型P0天高温10天光照10天高湿10天16.6019706.6548916.6259266.57413528.1554738.2169638.1358998.152156310.87070010.82632510.92126310.895623411.88593011.84135811.93162511.862592512.95490012.89269212.93159213.024856614.97511014.94586214.98235615.025889715.58157015.55826815.62389315.581662820.26818020.31693220.24581120.285583923.82968023.87166923.79494623.861449表5晶型Q在高温、高湿、光照条件下的粉末衍射角度数据(2θ衍射角)PeakNo.晶型Q0天高温10天光照10天高湿10天16.4836226.5488566.5216356.45592326.7008186.6516986.7165526.745691310.19969010.23491610.18592110.158916412.29944012.27156112.35645612.321583512.95873013.01591612.98151412.921162613.38755013.42159213.35846113.378141714.98096015.04789514.94546814.994681821.32225021.34581221.27861821.371592924.84981024.79512324.82458624.871686表6晶型T在高温、高湿、光照条件下的粉末衍射角度数据(2θ衍射角)PeakNo.晶型T0天高温10天光照10天高湿10天111.28811011.32589611.31585911.294583211.83385011.82489311.79266111.814692315.94038015.98145615.95482615.895492416.54999016.57489216.49826216.582262518.91151018.94856218.88926218.894825619.28069019.24861319.31565819.325491719.74457019.78156119.71459219.754816821.79384021.84591221.83149121.768911923.67941023.68161323.72146423.644513表7晶型X在高温、高湿、光照条件下的粉末衍射角度数据(2θ衍射角)试验例4吸潮性考察试验将实施例1制备的无定型、实施例2~10制备的晶型B、M1、O、P、Q、T、U、V和X样品分别放入洁净的培养皿中敞口平摊放置,考察在常温常湿(25℃、RH50%)条件下放置,检查其质量增加百分比,结果如下表8所示:表8吸湿性试验批号晶型6h12h24h48h20140309无定型0.2%0.4%0.7%0.9%20140408晶型B0.01%0.03%0.05%0.08%20140409晶型M10.3%0.5%0.6%0.7%20140414晶型O0.2%0.5%0.7%0.9%20140415晶型P0.3%0.5%0.7%0.9%20140418晶型Q0.2%0.4%0.5%0.7%20140421晶型T0.3%0.7%1.2%1.8%20140424晶型U5.6%7.7%9.1%9.4%20140425晶型V0.3%0.6%0.8%1.0%20140427晶型X0.02%0.04%0.07%0.09%试验结果表明,在常温常湿(25℃、RH50%)条件下,本发明式I化合物的晶型B、晶型X,质量增加百分比均小于0.2%,即无吸湿性;晶型M1、晶型O、晶型P、晶型Q、晶型T、晶型V质量增加百分比在0.2%-2%范围内,即略有吸湿性;晶型U质量增加百分比大于2%范围内,即有吸湿性。效果例式I化合物及其各晶型对正常小鼠APTT的作用试验试验目的:考察5mg/kg给药剂量下各实施例制备的晶型在给药后对小鼠APTT的作用。1.试验材料:1.1.药品:受试药:式I化合物(无定型),实施例1制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140309;阳性对照药:阿哌沙班,购自上海皓元化学科技有限公司,纯度99.9%,批号HM-038_13-20140427;受试药:晶型B,实施例2制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140408;受试药:晶型M1,实施例3制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140409;受试药:晶型O,实施例4制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140414;受试药:晶型P,实施例5制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140415;受试药:晶型Q,实施例6制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140418;受试药:晶型T,实施例7制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140421;受试药:晶型U,实施例8制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140424;受试药:晶型V,实施例9制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140425;受试药:晶型X,实施例10制备,由成都苑东药业有限公司合成研究室提供,浅黄色固体,批号:20140427;1.2试验器材:SysmexCA7000型全自动血凝分析仪;RocheC501全自动生化分析仪;采血管、手术剪、注射器等;1.3试验动物:KM小鼠,体重22~24g,120只,雌雄各半,由成都达硕生物科技有限公司提供,生产设施许可证:SCXK(川)2013-24。动物购回后饲养于动物房,适应性观察至少3天,检疫合格后用于实验。2.1分组:小鼠根据体重按照表9分组,每组10只,雌雄各半,组间无统计学差异;表9试验分组及给药方案2.2APTT测定:按照表9分别对各组灌胃给予相应受试药(空白组给予生理盐水),给药1h后,眼眶取血于含枸橼酸钠的0.5ml真空采血管中,采集血样后测定各动物的APTT;3.统计学方法:采用Excel进行统计,实验数据采用表示,多组之间比较采用双侧T检验方法进行统计学比较。4.试验结果:表10对正常小鼠APTT的影响组别受试物给药剂量(mg/kg)APTT值(s)空白组生理盐水516.11±1.05阳性组阿哌沙班521.58±0.97**实施例1组无定型527.92±2.65**△△实施例2组晶型B531.42±2.53**△△◇◇实施例3组晶型M1530.58±2.40**△△◇实施例4组晶型O530.78±2.18**△△◇实施例5组晶型P529.07±2.91**△△实施例6组晶型Q530.06±1.73**△△◇实施例7组晶型T528.97±1.84**△△实施例8组晶型U528.54±3.70**△△实施例9组晶型V529.88±2.93**△△实施例10组晶型X531.00±2.91**△△◇注:与空白组相比*P<0.05;**P<0.01;与阳性组相比△P<0.05;△△P<0.01;与实施例1组相比◇P<0.05;◇◇P<0.01。5.结论:(1)从表10中可以看出,与空白组相比,给药1h后,阳性组、各实施例组的APTT(活化部分凝血活酶时间)值极显著增加(P<0.01),说明阳性药阿哌沙班、实施例1及其各晶型在给药1h后均能极显著延长小鼠的APTT值;(2)与阳性组相比,各实施例组的APTT值极显著增加(P<0.01),说明实施例1化合物(无定型)及其各晶型对延长小鼠APTT值均优于阳性组;(3)与实施例1组相比,实施例2组的APTT值极显著增加(P<0.01)、而实施例3、4、6、10组的APTT值显著增加(P<0.05),说明实施例2化合物的晶型B、实施例3化合物的晶型M1、实施例4化合物晶型O、实施例6化合物晶型Q、实施例10化合物晶型X的效果相对更好。对于本领域的普通技术人员而言明显的是,在不偏离本发明精神或者范围的情况下,可对本发明化合物、组合物以及其制备方法进行的多种修饰和变化,因此,本发明的保护范围涵盖了对本发明进行的各种修饰和变化,只要所述修饰或变化处于权利要求和其等同实施方式所涵盖的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1