TALENs介导的奶山羊成纤维细胞基因敲除和基因定点插入的方法与流程
本发明涉及分子生物学及生物技术领域,具体地说,涉及TALENs介导的奶山羊成纤维细胞基因敲除和基因定点插入的方法。
背景技术:
羊奶以其营养丰富、易于吸收等优点被视为乳品中的精品,被称为“奶中之王”,也因相较于牛奶,其小脂肪球,高乳清蛋白含量以及与人奶基本相同的蛋白质构成的特点,被世界公认为最接近人奶的乳品,更因为其含有较高含量的维生素,如A,C,烟酸,尼克酸等,是现代人类健康的营养佳品。另一方面,羊奶对于多数亚洲人由于肠道中缺乏乳糖酶而对牛奶乳糖产生的低过敏和不耐受有比较好的适应性。陕西省是全国奶山羊大省,具有著名的莎能奶山羊、关中奶山羊等优良品种,羊奶制品加工处于国内领先水平。
酪蛋白属磷蛋白类,是反刍类动物乳汁中主要的奶蛋白,约占乳汁蛋白含量的80%。β酪蛋白(β casein,CSN2)是羊乳酪蛋白成分中含量最多的一种酪蛋白,在羊奶加热沉淀中,蛋白质是其主要成分,尤其是羊奶蛋白质的主要成分-酪蛋白,羊奶酪蛋白热稳定性随温度的升高而降低,而有研究也表明其他物理、化学等条件的变化也都将影响酪蛋白稳定性,从而影响羊乳稳定性。不同品种奶山羊奶的酪蛋白热稳定性也有一定差别,莎能羊奶酪蛋白的热稳定性高于关中羊奶酪蛋白。
基因打靶技术是近年来发展起来的一种对细胞遗传信息进行定点的修饰、修改,或者将外源基因导入的基因组内特定位置的转基因技术。获得基因打靶的细胞后,再通过体细胞核移植技术和胚胎移植技术,可以最终获得转基因动物个体。这种方法能够在1~2个代次内获得具有稳定遗传性状的个体,大大提高了动物改良和选育的速度。传统的基因打靶技术效率约10-7,效率较低,并且需要经过多种手段的筛选,最终得到的细胞代次高,细胞状态差。近年来,人们发现了一种新的基因组编辑技术,即将识别特异DNA序列的TALE与核酸内切酶Fok I偶联,可构建成剪切特异DNA序列的内切酶TALEN(transcription activator-like effector nucleases)。而且Fok I需形成2聚体方能发挥活性,大大减少了随意剪切的几率。由转录激活因子样效应物核酸酶介导的基因剪切具有较高的效率,并且相较于传统的打靶技术,TALEN具有较高的同源重组效率,毒性低和低脱靶的特点,也克服了常规的ZFN方法不能识别任意目标基因序列,以及识别序列经常受上下游序列影响等问题,而具有ZFN相等或更好的活性。目前,该技术已经成功应用到了细胞、植物、酵母、斑马鱼等各类研究对象,以及哺乳动物细胞的基因修饰和转基因的实验研究中。而在针对奶山羊β-酪蛋白基因的敲除和打靶上尚未有报道。
鱼油里的长链不饱和脂肪酸ω-3已经被证实有益于人类健康,膳食结构中ω-6/ω-3不饱和脂肪酸的高比例是导致许多疾病如癌症、糖尿病、冠状动脉硬化的发生的主要原因之一。哺乳动物自身不能合成ω-3脂肪酸,而低等动物线虫中具有行使这种转化功能的fat-1基因。将线虫fat-1基因经人源化修饰后,转入小鼠和猪,结果转基因小鼠和猪的肌肉里的ω-3:ω-6的比率可以比野生型小鼠和猪的比率提高5倍。最近,有报道称成功培育了fat-1转基因奶牛,但fat-1转基奶山羊的研究上未见报道,如果能够获得这种将fat-1基因整合到β-酪蛋白基因中,实现在羊奶中高含量的fat-1表达,则能够改良羊奶中脂肪酸的比例,提高羊奶品质。
利用转录激活因子样效应物核酸酶敲除奶山羊β-酪蛋白基因,同时在转录激活因子样效应物核酸酶切割位点定点整合hfat-1基因的研究未见报道。
技术实现要素:
本发明的目的是提供一种TALENs介导的奶山羊成纤维细胞基因敲除和基因定点插入的方法。
为了实现本发明目的,本发明的TALENs介导的奶山羊成纤维细胞基因敲除和基因定点插入的方法,其是根据奶山羊CSN2基因序列,获得针对第二外显子的TALENs表达载体,并根据TALENs作用位点设计含有外源基因的且可以整合至宿主基因组中的打靶载体,然后将上述TALENs表达载体和打靶载体共同转入奶山羊成纤维细胞中,获得CSN2基因敲除且定点整合外源基因的细胞。其中,TALENs作用位点的核苷酸序列为5′-TCGAGAGCCATGAAGGTCCTCATCCTTGCCTGTCTGGTGGCTCTGGCCATTGCA-3′,即CSN2基因的第3745-3799bp,该作用位点位于CSN2基因的第二外显子内。
本发明提供的打靶载体含有根据TALENs作用位点设计的奶山羊CSN2基因5’同源臂和3’同源臂。
前述的方法,所述外源基因为经过人源化修饰后的fat-1基因。具体方法如下:
根据奶山羊CSN2基因序列,获得针对第二外显子的TALENs表达载体(gMK-L和gMK-R);其中TALENs作用位点的核苷酸序列为5′-TCGAGAGCCATGAAGGTCCTCATCCTTGCCTGTCTGGTGGCTCTGGCCATTGCA-3′;
2)根据TALENs作用位点构建含有外源基因的且可以整合至宿主基因组中的打靶载体pflrk,其核苷酸序列如SEQ ID NO:1所示;
3)将上述表达载体和打靶载体共同转入奶山羊成纤维细胞中,通过PCR产物测序检测CSN2基因敲除且定点整合外源基因的细胞。
本发明还提供用于TALENs介导的奶山羊成纤维细胞基因敲除和基因定点插入的载体,所述载体包括针对奶山羊CSN2基因第二外显子序列设计的TALENs表达载体,以及根据TALENs作用位点设计含有外源基因的且可以整合至宿主基因组中的打靶载体。
本发明还提供由上述方法获得的CSN2基因敲除且定点整合外源基因的细胞。
本发明还提供上述方法在生产CSN2基因敲除且定点整合外源基因的克隆奶山羊中的应用。
本发明还提供一种制备CSN2基因敲除且定点整合外源基因的奶山羊克隆胚胎的方法,其以所述CSN2基因敲除且定点整合外源基因的细胞为核移植供体细胞,离体的卵母细胞为核移植受体细胞,通过核移植技术获得奶山羊克隆胚胎。
本发明还提供一种制备转基因奶山羊的方法,其是将按上述方法制备的克隆胚胎通过非手术法移入奶山羊子宫内进行妊娠,获得转基因奶山羊。
本发明的目的还可以采用以下的技术措施来进一步实现。
方案I:提供针对奶山羊β-酪蛋白第二外显子的转录激活因子样效应物核酸酶载体,对β-酪蛋白基因进行突变,达到β-酪蛋白的失活。具体如下:
从GenBank获得β-酪蛋白全长DNA序列(GenBank:AJ011019.3),针对第二外显子设计一套转录激活因子样效应物核酸酶载体,即TALENs表达载体,命名为gMK-L(SEQ ID NO:9)和gMK-R(SEQ ID NO:10),通过电转染的方法,将转录激活因子样效应物核酸酶载体导入奶山羊成纤维细胞中,再挑取单细胞建立多个单细胞系,将同一细胞系分为两部分,分别传代和冻存,对传代的细胞系提取基因组,使用跨作用位点的PCR检测引物进行扩增,扩增产物进行测序鉴定,最终得到在转录激活因子样效应物核酸酶作用位点发生突变细胞株。
方案II:提供针对方案I中的转录激活因子样效应物核酸酶作用位点的打靶载体,将外源基因hfat-1靶向整合到转录激活因子样效应物核酸酶的靶向位点。具体如下:
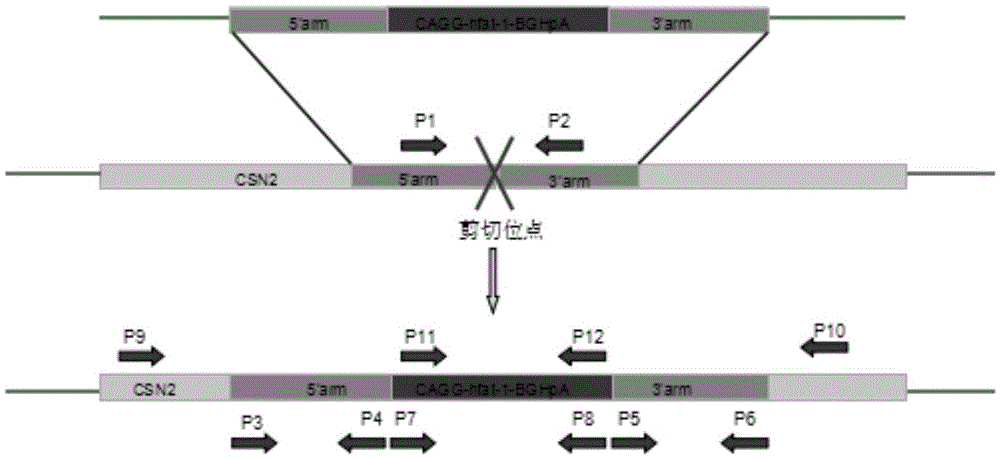
从奶山羊成纤维细胞获得基因组DNA,根据GenBank中公开的β-酪蛋白全长序列,分别设计针对转录激活因子样效应物核酸酶作用位点的同源臂引物,通过PCR扩增出同源臂,通过连接克隆载体并进行限制酶切和测序鉴定为正确序列后,采用酶切和连接的方法将5’同源臂和3’同源臂分别连接到基础载体pCAGG-hfat-1-BGHpA的预期位置,最后通过限制酶酶切鉴定同源臂的正确性,最终获得针对TALENs作用位点的打靶载体pflrk(构建过程见图1和图2)。
本发明提供的打靶载体pflrk是针对TALENs作用位点的打靶载体,以pCAGG-hfat-1-BGHpA为基础载体,包括CAGG启动子下的人源化fat-1基因和PGK启动子下的新霉素抗性基因(Neor),以及新霉素抗性表达框架两侧的LoxP位点。根据TALENs作用位点的上下游序列,在CAGG启动子上游插入长度为1018bp的5’同源臂,位于CSN2序列的2736-3754,在Neor表达框架下游包含长1022bp的3’同源臂,位于CSN2序列的4312-5334。同源臂序列位于TALENs作用位点的两侧。当pflrk与gMK-L和gMK-R共转染细胞时,gMK-L和gMK-R在识别位点发生剪切,经同源重组作用,基因组以pflrk为模板,将外源基因靶向整合到目的区域。
上述pflrk载体的同源臂引物,包括5’同源臂和3’同源臂的上游引物和下游引物两组。具体引物序列如下(5′-3′):
5’同源臂上游引物:LH-1U:ACGCGTTTTTCAAGAACTTATAAAAATGC
5’同源臂下游引物:LH-1D:ACGCGTGGCTCTCGATTCCTGTGAATGG
3’同源臂上游引物:RH-1U:GCTAGCAAATAACTCCTTTTCTGTTAGC
3’同源臂下游引物:RH-1D:GCTAGCCTGCCCACACTTTACAAAGTGC
具体地,本发明提供的β-酪蛋白基因敲除方法包括以下步骤:
1、奶山羊成纤维细胞的培养;
2、利用电转染的方法将gMK-L和gMK-R转染到步骤1中培养的奶山羊成纤维细胞中;
3、利用直径约10μm的口吸管,从步骤2中转染的奶山羊成纤维细胞中挑取细胞单克隆并传代培养,提取基因组;
4、利用PCR方法对步骤3中的基因组进行特异性扩增,通过测序的方法鉴定得到阳性敲除细胞;
5、对步骤4中获取的阳性敲除细胞进行扩大培养,并进一步利用鉴定引物PCR扩增,连接载体后进行测序和比对分析,最终获得突变的详细信息。
其中步骤4中所述的PCR引物序列(5′-3′)为:
突变检测上游引物:61:CGCTGTTTCCTCATCTTCCCA
突变检测下游引物:62:TTACCTCACTGCTTGAAAGGC
PCR反应条件为:95℃变性30s,60℃退火30s,72℃延伸30s,共35个循环。
本发明提供的针对TALENs作用位点的打靶载体pflrk的主要构建步骤如下:
1、提取奶山羊成纤维细胞基因组DNA;
2、利用引物LH-1U和LH-1D,从步骤1中的基因组DNA中克隆β-酪蛋白的5’同源臂,利用引物RH-1U和RH-1D从步骤1中的基因组DNA中克隆β-酪蛋白的3’同源臂;
3、将同源臂连接到克隆载体中,进行限制性内切酶分析和测序分析鉴定;
4、将鉴定正确的同源臂连接到基础载体中,构建得到针对TALENs作用位点的打靶载体pflrk。
其中步骤2中所述的PCR方法扩增5’同源臂的反应条件为:95℃变性30s,60℃退火30s,72℃延伸2min,共35个循环;3’同源臂的反应条件为:95℃变性30s,60℃退火30s,72℃延伸2min,共35个循环。
G418(Geneticin selective antibiotic 418)是一种抗生素类药物,当Neor基因进入细胞后,细胞获得抗性,而在G418培养基中可以生长,而未获得抗性的细胞则在G418培养基中死亡。G418这一特性已在转基因技术、基因敲除等方面广泛应用。
本发明提供的基因敲除方法,利用电转染方法,将gMK-L和gMK-R导入到奶山羊成纤维细胞中,进入细胞后的载体分别表达转录激活因子样效应物核酸酶,转录激活因子样效应物核酸酶由特异性识别DNA序列的TAL结构域和非特异性的DNA剪切结构域两部分构成,转录激活因子样效应物核酸酶能够在细胞中特异性识别靶向位点,并在该位点将DNA剪切,形成双链断裂,并启动DNA的自我修复机制。DNA可以通过两种不同的途径修复,第一种为同源重组,损伤的DNA可以以等位基因为模板,修复DNA损伤,这种修复方式产生的DNA与原来的没有区别,不会发生突变;另一种为非同源性末端修复,损伤的DNA不以其他等位基因为模板,随机对断裂的DNA进行修复,这种修复方式会产生DNA的碱基缺失或插入突变,最终造成基因的突变。gMK-L和gMK-R针对β酪蛋白基因的第二外显子的序列,能够在其针对位点产生DNA双链断裂,引起DNA的修复,而产生突变。对转染后的细胞用口吸管挑取单细胞,分别接种在96孔板中,经过传代和提取基因组,并进行PCR扩增和测序分析,最终得到发生突变的细胞系。
本发明还提供一种基因打靶的方法,利用电转染方法,分别将gMK-L和gMK-R以及pflrk共同导入到奶山羊成纤维细胞中,转录激活因子样效应物核酸酶在靶向位点对DNA进行切割产生双链断裂,基因组在进行同源修复时,以打靶载体为模板将外源基因整合到靶向位点。外源基因带有Neor,使细胞在含有G418的培养液中存活,在经过G418的筛选后,挑取细胞单克隆,经过PCR检测和测序的方法,得到打靶的细胞系。同时整合了外源基因的细胞,β酪蛋白基因由于外源基因的插入,不能正确表达β酪蛋白,同时达到敲除β酪蛋白基因的目的。
本发明进一步提供了转录激活因子样效应物核酸酶介导的基因敲除和基因打靶在培育奶山羊品种中的应用。
本发明具有以下优点:
(一)本发明首次利用TALENs技术实现奶山羊β酪蛋白基因的敲除,TALENs技术具有实验设计简单准确、实验周期短、成本低、成功率高、脱靶情况少等优点。利用本方法可以快速获得双等位基因整合的细胞克隆,细胞不经过药物筛选,提高了所获得体细胞的核移植和胚胎发育质量,不含有抗性基因,大大简化了生物安全评价。
(二)奶山羊β酪蛋白基因的成功敲除利于后期生物反应器的制备,通过改进山羊奶的品质,提高了经济效益。
附图说明
图1为本发明打靶载体pflrk构建流程图。
图2为本发明TALENs介导的基因打靶过程。
图3为本发明实施例4中同源臂连接T载体后酶切图;其中,1-4为pMD-19T载体连接5’同源臂后获得的重组质粒酶切结果;2、3有明显的小片段,是阳性重组质粒;5-8为pMD-19T载体连接3’同源臂后获得的重组质粒酶切结果;6、7泳道有小片段产生,是阳性重组质粒;9为空载体酶切结果。
图4为本发明实施例5中间载体pfr酶切鉴定图;其中,M1为DNA分子量标准1000bp ladder(Takara公司);1-3为来自3个不同菌落的pfr质粒酶切结果,产生大小为124bp、8932bp的片段,为同源臂正向连接结果。
图5为本发明实施例6中打靶载体pflrk酶切图;其中,M1为DNA分子量标准1000bp ladder(Takara公司);1-7为来自7个不同菌落的pflrk质粒酶切结果,其中1-3为3867bp和6320bp酶切条带,说明质粒正确,4-7产生大小为4096bp和5991bp的酶切片段,说明质粒不正确。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例均按照常规实验条件,如Sambrook等分子克隆实验手册(Sambrook J&Russell DW,Molecular cloning:a laboratory manual,2001),或按照制造厂商说明书建议的条件。
实施例1 奶山羊成纤维细胞基因组的提取
将培养于100mm培养皿中的细胞消化后,离心收集,用Promega公司的Wizard Genomic DNA Purification Kit试剂盒,按照提取细胞基因组的方法,进行奶山羊基因组DNA提取,最后用100μL超纯水回溶DNA。利用紫外分光光度计检测提取DNA的浓度和纯度,用于后续工作。
实施例2 TALENs表达载体gMK-L和gMK-R的构建
根据奶山羊CSN2基因序列,获得针对第二外显子的TALENs表达载体gMK-L和gMK-R。其中,TALENs作用位点的核苷酸序列为5′-TCGAGAGCCATGAAGGTCCTCATCCTTGCCTGTCTGGTGGCTCTGGCCATTGCA-3′。
TALENs表达载体的构建方法简述如下:
针对奶山羊CSN2基因序列设计TALENs序列。然后使用LR重组技术,将表达载体和TALENs序列载体重组得到最终的TALEMs表达载体gMK-L(SEQ ID NO:9)和gMK-R(SEQ ID NO:10)。
实施例3 β酪蛋白基因同源臂pMD19-T-LA-1和pMD19-T-RA-1的获取
根据奶山羊CSN2基因序列(GenBank:AJ011019.3),分别设计针对TALENs作用位点的打靶载体的5’同源臂的上游引物LH-1U(ACGCGTTTTTCAAGAACTTATAAAAATGC)和下游引物LH-1D(ACGCGTGGCTCTCGATTCCTGTGAATGG);针对TALENs作用位点的打靶载体的3’同源臂的上游引物RH-1U(GCTAGCAAATAACTCCTTTTCTGTTAGC)和下游引物RH-1D(GCTAGCCTGCCCACACTTTACAAAGTGC)下划线部分为酶切位点。以上述羊基因组DNA为模板,分别进行PCR合成同源臂。
PCR反应为20μL体系:上下游引物各1μL,模板1μL,10×反应缓冲液2μL,2.5mM dNTPs 2μL,EX-Taq 0.5μL,ddH2O 10μL。针对TALENs作用位点的打靶载体的5’同源臂PCR反应条件为:95℃变性30s,60℃退火30s,72℃延伸2min,共35个循环;3’同源臂的反应条件为:95℃变性30s,60℃退火30s,72℃延伸2min,共35个循环。
将扩增后得到的PCR产物分别连接到pMD19-T载体(Takara公司)上,转化E.coli DH5α,分别挑取阳性连接菌落进行扩培和质粒提取,最终获得pMD19-T-LA-1和pMD19-T-RA-1两种同源臂质粒。
实施例4 同源臂的酶切鉴定和测序分析
对两种同源臂质粒分别取1μL,利用限制性内切酶EcoRI、XbaI和BfaI进行酶切鉴定,左右同源臂质粒的同源臂上分别有XbaI和BfaI酶切位点且两者酶切位点相近,EcoRI位于pMD19-T载体的多克隆位点中,可以初步检测PCR获得的同源臂的正确性(图3)。酶切体系均为10μL:同源臂质粒1μL,10×酶切反应缓冲液1μL,内切酶0.5μL,ddH2O 7.5μL。将酶切鉴定正确的质粒进行DNA测序分析,并与GenBank中的奶山羊β酪蛋白基因序列比对,最终确定同源臂序列的正确性。
实施例5 3’同源臂连入基础载体
将测序正确的同源臂质粒pMD19-T-RA-1及基础载体pCAGG-hfat-1-BGHpA,载体pCAGG-hfat-1-BGHpA的构建过程参照《人凝血因子IX乳腺特异表达载体的构建及细胞转染》,韩雪洁等,2014。分别用限制性内切酶NheI进行酶切,酶切体系如前,分别回收pMD19-T-RA-1的1022bp(1Kb)片段,以及骨架载体的线性化片段,然后将pMD19-T-RA-1与pCAGG-hfat-1-BGHpA按照摩尔比2:1的比例在16℃条件下,经T4DNA连接酶过夜连接,转化E.coli,提取质粒分别命名为pfr。利用限制性内切酶HpaI和BstBI酶切鉴定连接的方向和正确性(图4),并最终确定3’同源臂正确连入基础载体。
实施例6 5’同源臂连入基础载体
将测序正确的pMD19-T-LA-1及中间载体pfr分别用限制性内切酶NheI进行酶切,酶切体系如前,分别回收pMD19-T-LA-1的1018bp(1kb)片段,以及中间载体pfr的线性化片段,然后将pMD19-T-LA-1与中间载体pfr按照摩尔比2:1的比例在16℃条件下,经T4DNA连接酶过夜连接,转化E.coli,提取质粒命名为pflrk。利用限制性内切酶BfaI和HindIII酶切鉴定连接的方向和正确性(图5),并最终确定5’同源臂正确连入载体。最终成功获得针对于TALENs作用位点的打靶载体。构建的打靶载体pflrk(图1),其核苷酸序列如SEQ ID NO:1所示。
实施例7 TALENs表达载体gMK-L、gMK-R和打靶载体pflrk共转染奶山羊成纤维细胞
将奶山羊成纤维细胞于38.5℃,5%CO2条件的细胞培养箱中培养传代24小时后用于电转染,消化离心,Opti-MEM清洗并稀释为1×104个/μL。首先将1000ng质粒DNA(gMK-L 500ng+gMK-R 500ng+pflrk 500ng)用无酶水稀释至10μL,再加入90μL奶山羊成纤维细胞,轻轻混匀后转移至点击杯中,以225V,2.5ms脉冲,脉冲间隔50ms,2次脉冲,10%电压衰减的条件下进行电转。电转染后的细胞接种于60mm培养皿中,用DMEM/F12+10%FBS培养液培养48小时后,将细胞消化重悬于PBS中,于体视显微镜下用自制口吸管吸取单个细胞,分别接种于含200μL培养液/孔的96孔板中培养,并于第3、5和8天对培养板换液。在第10天时将培养板中生长的细胞单克隆传代至24孔板中,继续培养3-5天后,将细胞单克隆一部分冻存,另一部分提取基因组进行鉴定。
以基因组染色体定点整合序列为模板,分别设计跨越3’和5’同源臂序列的PCR引物,利用PCR法和DNA测序检测同源重组事件。PCR方法所用的引物序列如下(5′-3′):
5fat-FWD:TAATGGATTCTAGGTATTATGC
5fat-REV:CACACAAGCAGGGAGCAGATAC
3fat-FWD:CTTGCCGAATATCATGGTGG
3fat-REV:AGGACTACGCTCATTCTCACTGC
5’同源臂PCR反应条件为:95℃变性30s,60℃退火30s,72℃延伸2min,共35个循环;3’同源臂PCR反应条件为:95℃变性30s,55℃退火30s,72℃延伸2min,共35个循环。
当待测序列同时包含MSTN基因5’同源臂上游序列、5’同源臂序列、3’同源臂下游序列、3’同源臂序列以及hfat-1基因时,则能够扩增出目的条带,经过测序检测正确后,则判定该细胞是中靶细胞,外源基因hfat-1成功整合到靶向位点。将该细胞作为体细胞核移植的供体,进行牛体细胞克隆及胚胎移植,最终获得MSTN基因敲除,并整合表达hfat-1基因的转基因动物。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
参考文献:
韩雪洁,萨日娜,梁浩,李雪玲,李荣凤。人凝血因子IX乳腺特异表达载体的构建及细胞转染。中国农业科学,2014,47(4):769-778。
- 还没有人留言评论。精彩留言会获得点赞!
- 用CRISPR/Cas9技术易于筛选获得目的基因敲除细胞系的方法及产品与制造工艺
- 一种Candida amazonensis的FLP/FRT基因敲除方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 利用CRISPR/Cas9敲除人PD‑1基因构建靶向CD19CAR‑T细胞的方法与制造工艺
- 一种构建基因替代小鼠研究野生型和突变体非肌性肌球蛋白重链II功能的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 猪ApoE基因敲除打靶载体及其构建方法和应用与制造工艺
- 一种血小板减少症的斑马鱼模型的制造方法与工艺
- 基于CRISPR技术敲除FUT8基因的方法与制造工艺